1. Introduction
In all computational branches of scientific research (Physics, Chemistry, Philosophy …) programs and computers play a central role. The programs used can be either locally produced code or commercial software. The latter is often used as a black box application. However, even when using software as a black box tool, it is important to have some knowledge of the limitations and quirks of the software used. These are obtained in two ways: firstly by studying (and trying to understand) the underlying theories and algorithms used, and secondly by experience. One will often discover that knowing how a code should behave, and how it actually does, can be quite different things (and “just checking out the code to see how it works”, is often easier said than done, when such code contains tens of thousands of lines of generally undocumented code).
As with most projects in science, also computational scientists want to trace the borders of what is possible, and if possible look beyond. For computational problems, there are two main computational resources that play a role:(1) time and (2) memory. As the problem size grows, so do these resources. The latter is mainly remedied by using machines with more RAM-memory (or through storing intermediate data on disc, which tend to be tremendously slow), the former can be remedied through parallel usage of multiple CPU’s. This is where supercomputer infrastructures come into play. Although a desktop machine can be useful for testing purposes, or small problems, a computational scientist also needs to be familiar with basic usage of parallel codes on a supercomputer. Actually, even modern day personal machines have a multi-core architecture (try to find a new computer anno 2014 with less than 4 cores) making parallelism also here an important concept.
2. Supercomputers
Most supercomputers run some version of a unix based OS, with all peculiarities involved. The extent of this installation can vary significantly, which will have its influence on the user experience (bare systems which only contain the most basic commands and programs installed may be considered efficient, but they tend to be rather annoying than useful. And no, vi is not something some-one in his right mind should use for text-editing;). Knowing how to work in the command-line-only environment of a supercomputer is, as such, a must for almost any computational scientist (of course there are exceptions).
In addition to the above, a computational scientist should also have a good knowledge of how efficient the code he/she uses is parallelized, meaning, he/she should do scaling tests. Having run VASP on several supercomputers (going from local clusters to national HPC facilities) over the years, I compiled useful information on the efficiency of the VASP code:
- CMS group cluster at University Twente (nl)
- The Aster, Teras and Huygens supercomputers at SARA (nl)
- The Stevin supercomputers at Ugent (be). On several of these machines I worked as pilot-user.
- The Flemish TIER-1 supercomputer (muk) located at Ugent (be). On this machine I was one of the pilot users, and I was granted calculation time for several projects over the years.
3. Code-development
3.1. HIVE-STM
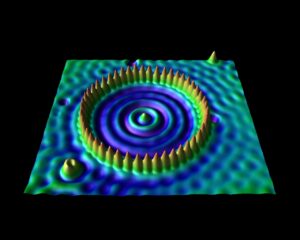
This is a Windows program written in Delphi that allows the user to generate simulated STM images based on VASP calculations. This program is freely-available to those interested. More information can be found on this page.
3.2. HIVE-3.x (personal development version)
This is the multi-purpose fortran-95/2003 program containing several subroutines to handle VASP output data. At the moment of writing the code counts over 20.000 lines of code and more than 30 command-line options. These vary from simple operations on POSCAR files(making supercells and surfaces), over the calculation of the vibrational contributions from phonon calculations, and fitting of E(V) data to different equations of state, to a full Hirshfeld and Hirshfeld-I atoms-in-molecules population analysis. Of course it also contains tools to extract DOS and band structure data from dedicated VASP calculations and present them in formats easily visualized with the xmgrace-software. Currently this software is not freely available. However, if you have access to the Ghent HPC facilities, you can use a beta-version of this program by simply loading the HIVE module.
3.2.bis. HIVE-4.x (personal development version)
This is a cleaned up version of HIVE 3.x which will become available for academic users. The program will be distributed as executable. More information can be found here.
3.3. Other Software
My interest in programming is not limited to solid state physics, or physics in general. Another subject which strongly draws my interest is fractals. Even though the algorithms have a limited size, they allow for the generation of very complex structures: e.g. fractal trees. In 2010 I spend some time writing a program for the generation of fractal trees and cities for a project of Dr. Yannick Joye.
Somewhat similar is my interest in the simulation of group “behavior” (note that from the physicists point of view a particle and a person are the same, as long as their behavior is described by rules that can be implemented). Again, from simple rules quite complex behavior can emerge, even though the overall behavior can still appear simple. In collaboration with Prof. Dr. Sylvia Wenmackers I have been working on a program to numerically calculate the chance for an agent to change its theory of the world to an inconsistent theory. (cf. Probability of inconsistency by theory updating under bounded confidence) The implementation of the algorithm allows us to find the exact analytic solution. However, the memory requirements and calculation time grows so fast with the problem size, that one quickly needs to move to a statistical approach, which is also implemented in our program.