

| Authors: | Danny E. P. Vanpoucke |
| Journal: | Computational Materials Science 181, 109736 (2020) |
| doi: | 10.1016/j.commatsci.2020.109736 |
| IF(2019): | 2.863 |
| export: | bibtex |
| pdf: | <ComputMaterSci> (Open Access) |
| github: | <Hive-toolbox> |
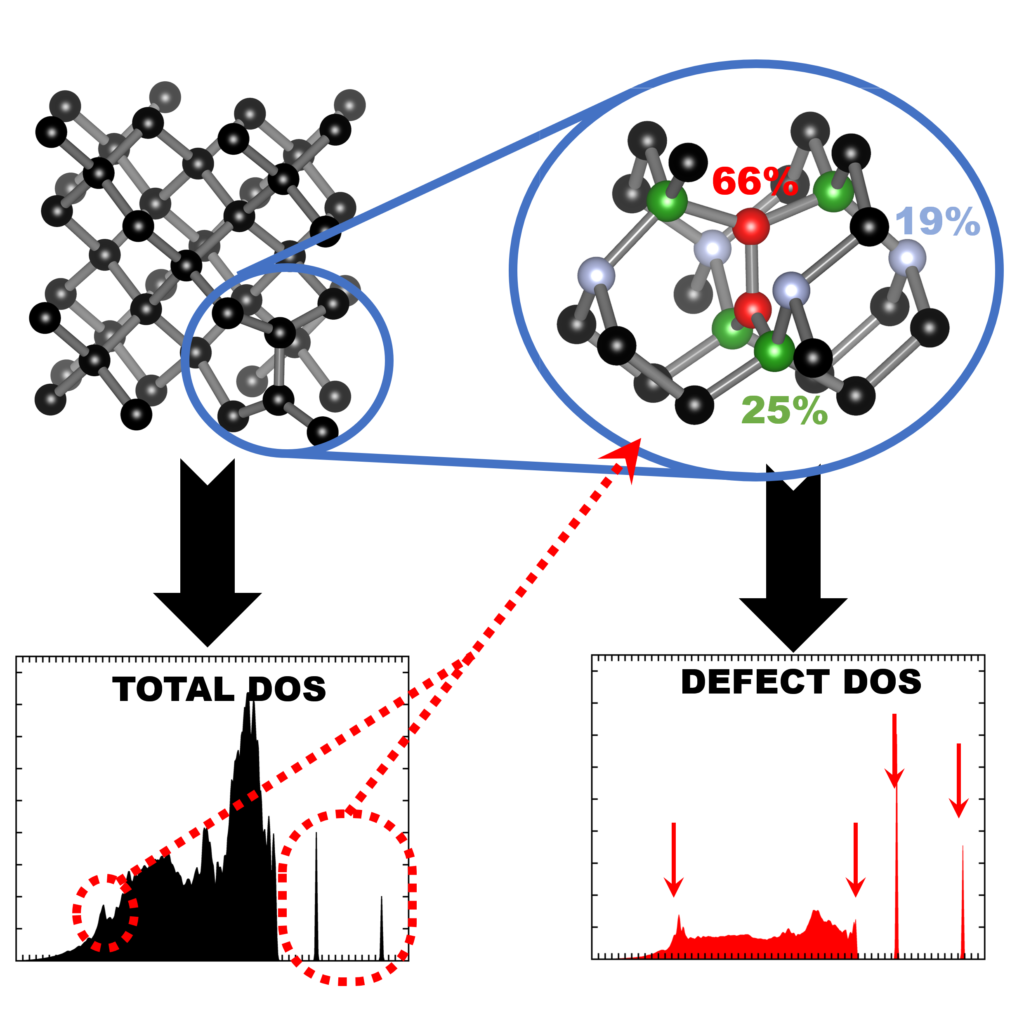 |
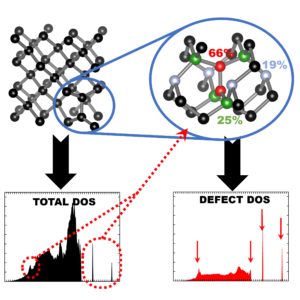
| Graphical Abstract: Finger printing defects in diamond through the creation of the vibrational spectrum of a defect. |
Abstract
Vibrational spectroscopy techniques are some of the most-used tools for materials
characterization. Their simulation is therefore of significant interest, but commonly
performed using low cost approximate computational methods, such as force-fields.
Highly accurate quantum-mechanical methods, on the other hand are generally only used
in the context of molecules or small unit cell solids. For extended solid systems,
such as defects, the computational cost of plane wave based quantum mechanical simulations
remains prohibitive for routine calculations. In this work, we present a computational scheme
for isolating the vibrational spectrum of a defect in a solid. By quantifying the defect character
of the atom-projected vibrational spectra, the contributing atoms are identified and the strength
of their contribution determined. This method could be used to systematically improve phonon
fragment calculations. More interestingly, using the atom-projected vibrational spectra of the
defect atoms directly, it is possible to obtain a well-converged defect spectrum at lower
computational cost, which also incorporates the host-lattice interactions. Using diamond as
the host material, four point-defect test cases, each presenting a distinctly different
vibrational behaviour, are considered: a heavy substitutional dopant (Eu), two intrinsic
point-defects (neutral vacancy and split interstitial), and the negatively charged N-vacancy
center. The heavy dopant and split interstitial present localized modes at low and high
frequencies, respectively, showing little overlap with the host spectrum. In contrast, the
neutral vacancy and the N-vacancy center show a broad contribution to the upper spectral range
of the host spectrum, making them challenging to extract. Independent of the vibrational behaviour,
the main atoms contributing to the defect spectrum can be clearly identified. Recombination of
their atom-projected spectra results in the isolated spectrum of the point-defect.

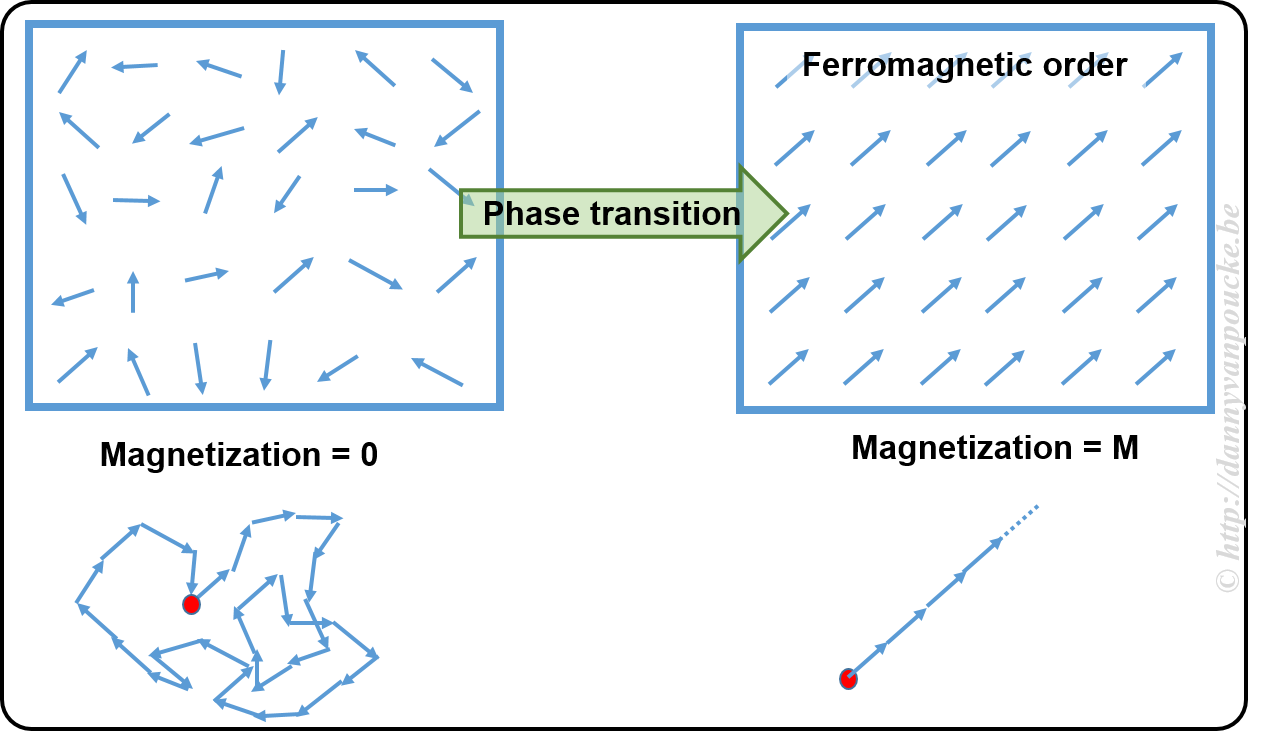
 A new year, a new beginning.
A new year, a new beginning.
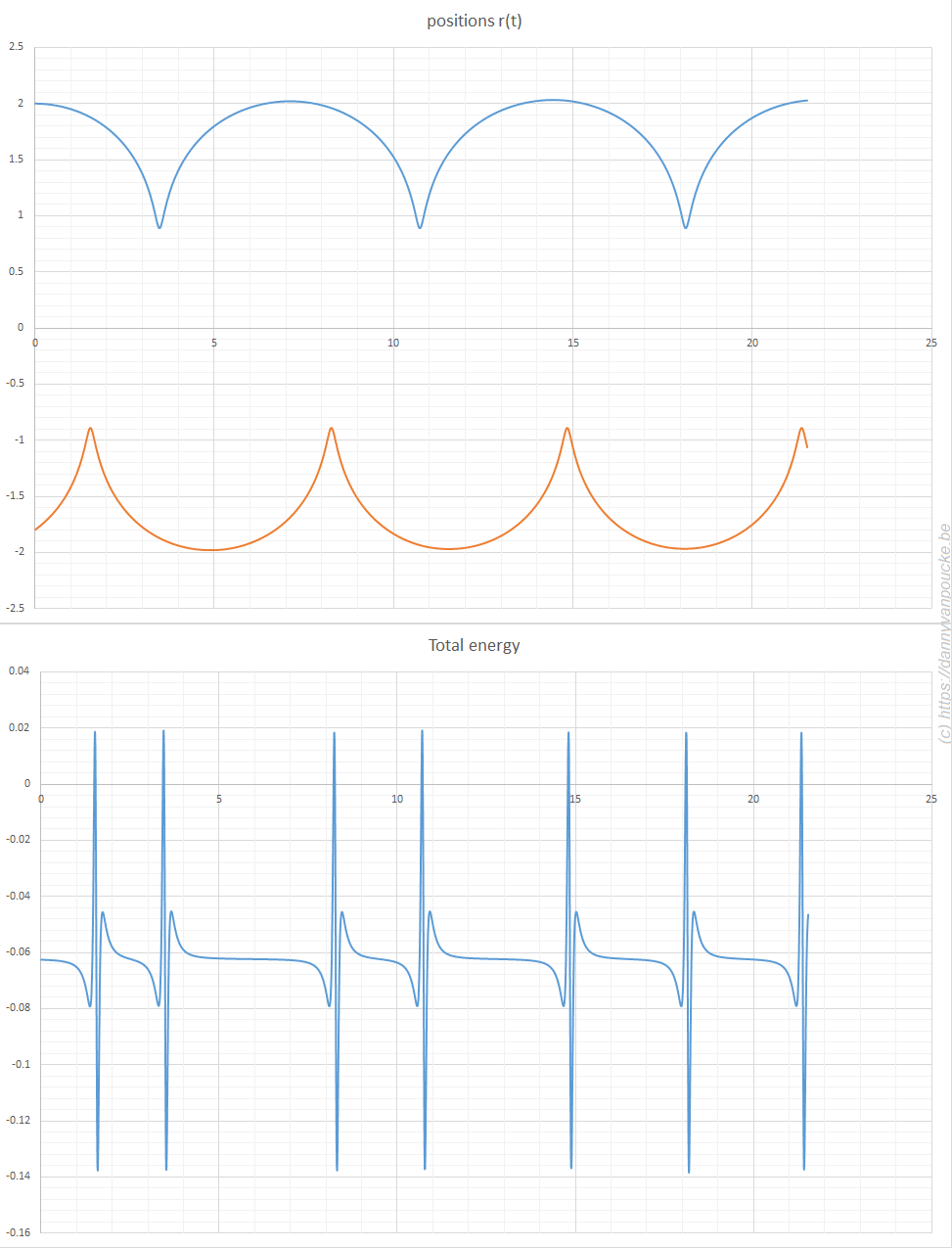






=-2/3(1-(1-3/2|x|)^{2/3})^{3/2}") ). When considering a point mass, experiencing only gravitational force, there are two solutions for the equation of motion: (1) the mass is there, and remains there forever (r(t)=0) and (2) the mass was rolling uphill with a non-zero speed which becomes exactly zero at the top, and continues over the top (
). When considering a point mass, experiencing only gravitational force, there are two solutions for the equation of motion: (1) the mass is there, and remains there forever (r(t)=0) and (2) the mass was rolling uphill with a non-zero speed which becomes exactly zero at the top, and continues over the top ( =\frac{1}{144} (t-T)^4") with T the time the top is reached). Here, r refers to the arc length as measured along the dome (0 at the top). In addition, there also exists a family of solutions taking the first solution at t<T, while taking the second solution at t>T. (As the first and second derivatives of these latter solutions are continuous, Newton will not complain.) This leads to indeterminism in a Newtonian system; for instance, you start with a mass on the top of the hill, and at a random point in time it starts to roll off without the presence of an external something putting it into motion. Using infinitesimals, Sylvia shows that the probability for the mass to start rolling off the dome immediately is infinitesimally close to one.
with T the time the top is reached). Here, r refers to the arc length as measured along the dome (0 at the top). In addition, there also exists a family of solutions taking the first solution at t<T, while taking the second solution at t>T. (As the first and second derivatives of these latter solutions are continuous, Newton will not complain.) This leads to indeterminism in a Newtonian system; for instance, you start with a mass on the top of the hill, and at a random point in time it starts to roll off without the presence of an external something putting it into motion. Using infinitesimals, Sylvia shows that the probability for the mass to start rolling off the dome immediately is infinitesimally close to one.