

1. Introduction
A major industrial application in the field of superconductivity is the production of superconducting wires, which in turn can be used to produce coils for superconducting magnets. In the development of high temperature superconducting wires and components, YBa2Cu3O7-δ remains one of the most important materials. Because it is a ceramic material, it is brittle by nature, making it ill-suited for wire-drawing. To produce an industrial scale wire with the required flexibility one can use a coated superconductor architecture instead. In such architecture, a thin film of the superconductor is deposited on a metal wire or tape, combining the flexibility of the metal substrate with the superconducting properties of the thin film. To prevent metal atoms of diffusing from the substrate into the superconductor, and thus destroying its superconducting properties, one or more buffer layers are deposited between the substrate and the thin film superconductor. Recently, cerium oxide (CeO2) has been used as such a buffer layer; however, the layer thickness of these buffer layers is limited by the formation of cracks during deposition, limiting their interest for industrial applications. Since economic factors demand the entire architecture to contain as few layers as possible, meaning that the individual buffer layers need to be a thick as possible, it is of great interest to increase the layer thickness before crack-formation starts. The formation of these microcracks has been linked to internal stress, and metal doping has been suggested as a means of reducing this internal stress, by alleviating the lattice mismatch and mismatch of the thermal expansion coefficients between the different layers.
In this work, we study the influence of dopants on the structural and mechanical properties of CeO2 using ab initio calculations. With the introduction of a dopant three separate changes take place in the system, each having its influence on the properties of CeO2. These are due to:
- Different electronic structure of the dopant and cerium.
- Different oxidation state of the dopant (aliovalent dopants) and cerium.
- As a consequence of point 2, the introduction of charge compensating oxygen vacancies.
To have a clear understanding of the influence dopants have on the properties of CeO2, it is important to separate the contributions of these changes; i.e. to distinguish between the consequences of doping (both the effect of the different electronic structure in case of isovalent dopants, and the effect of the oxidation state in case of aliovalent dopants) and subsequently those of the introduced charge compensating vacancies. Theoretical calculations are ideally suited to distinguish between these different contributions, since they allow the system under study to be tailored to these specific needs.
Although there exists an impressive body of theoretical work on doped CeO2; this work generally focuses either on a single aspect, of use for a single application, using a specific dopant, or investigates a property for a lanthanide series doped system. However, no general study of the influence of dopants on the properties of CeO2 exists. Such a study allows one to predict new possible dopants, but also provides ways to estimate required dopant concentrations for tuning applications (e.g. lattice parameter matching).
2. Vegard’s law and Shannon crystal radii
Cerium oxide is known to have a cubic fluorite crystal structure (cf. Fig. 1a). Assuming the bond length between two atoms to be the sum of their radii, it is possible to derive a linear relation between the lattice parameter of a doped CeO2 system and the dopant concentration, which is known as Vegard’s law:[2]
\big)")
where the ax are lattice parameters, Rx atomic radii and nx a concentration. This shows that for tetravalent dopants and aliovalent dopants under oxidizing conditions one should expect a linear relation between the dopant concentration and the lattice parameter. Although lattice parameters are experimentally available properties, atomic radii are not. It is true that these can be found in tables, but it is also true that different definitions used for atomic radii easily give rise to values which differ up to 20%. The question then becomes which definition one should use. In light of this problem it is of interest to note that the above equation can also be rewritten to yield the atomic radius of the dopant, as a function of lattice parameters and the atomic radius of oxygen.
Using this relation, we have calculated the atomic radius of the dopants under study and found the obtained values to be in good agreement with the tabulated Shannon crystal radii. As a consequence, this provides us with a simple and elegant method for deducing the oxidation state of a dopant in an experimental system. Using the measured lattice parameter, an atomic radius for the dopant can be calculated, and through comparison to the tabulated Shannon crystal radii, it is now possible to deduce the oxidation state of the dopant (under the assumption that the measurement was done under oxidizing conditions).
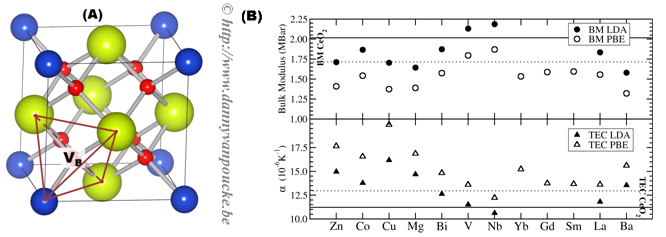
Figure 1. (a) Ball-and-stick representation of cubic fluorite CeO2 (Ce/O: red/yellow spheres) with 25% doping (blue spheres). An oxygen vacancy at a site VB is indicated, as is the surrounding tetrahedron. (b) Calculated bulk moduli (BM) and thermal expansion coefficients (TEC) at 500K for 25% doped CeO2.
3. Tetravalent dopants
Because cerium is tetravalent in cerium oxide, we focus on the group IV elements as dopants in a first step. Our study shows there exists a clear difference between the group IVa and group IVb elements, indicating that the specific electronic structure of the dopant (p-block compared to d-block) may be more important than the dopant oxidation state. We find that the group IVa elements are unstable dopants (unlikely to form uniformly doped CeO2) while the group IVb elements are stable dopants (rather likely to form uniformly doped CeO2). The calculated bulk moduli and thermal expansion coefficients show a similarly different behavior, where doping with a group IVa element lowers the bulk modulus, the introduction of group IVb elements slightly increases it, while the opposite is observed for the thermal expansion coefficients.[2,7]
4. Aliovalent dopants
The opposite behavior for the bulk modulus and the thermal expansion coefficients appears to be a general trend, which is also observed for aliovalent dopants with and without charge compensating oxygen vacancies (cf. Fig. 1b). Without the introduction of these vacancies, similar results are found for the aliovalent dopants as are found for the tetravalent dopants: Of the dopants investigated, the defect formation energies show Sm, Gd, La, and Nb to be the most favorable candidates for uniform doping of bulk CeO2. On the other hand, Zn, Co, and Cu are expected to be useful candidates for surface or interface doping, which is in agreement with experimental evidence.
In contrast to the group IV dopants, the calculated atomic radii of the aliovalent dopants are indicative of a coordination of less than 8, in turn pointing to the possibility that these dopants will introduce local deformations in the crystal lattice.
The introduction of charge compensating oxygen vacancies have a significant influence on all properties studied:[2,8]
- The doped system becomes slightly more stable (less unstable). However, this improvement of the defect formation energy is never so strong as to give a qualitatively different picture from the one obtained for aliovalent dopants without oxygen vacancies.
- The lattice parameter increases relative to the system without vacancies, compensating a lattice contraction or increasing a lattice expansion.
- The bulk modulus shows a very strong reduction, allowing for bulk modulus matching with La2Zr2O7 at low dopant concentrations, comparable to those required for bulk modulus matching (~5%).
5. Crystal structures of CeO2 based materials: La2Ce2O7 as example
A recurring aspect in the study of doped systems is the uncertainty of the exact atomic structure. Although the crystal structure for CeO2 is well established, this is not the general case for doped and mixed CeO2-based systems. An interesting case in this regard is lanthanum cerate (La2Ce2O7), despite the fact that this material was first produced and described in the late 1930’s by Zintl and Croatto, the ground state crystal structure remains a point of debate. The two proposed models are the pyrochlore structure and the disordered fluorite structure. This complicates the debate since both proposed structures are derived from the CeO2 fluorite crystal structure, so the resulting X-ray diffraction (XRD) and neutron diffraction spectra resemble the fluorite spectra very closely. As a result, both proposed models are supported by XRD and neutron diffraction measurements which either observe or don’t observe certain reflections ascribed to the pyrochlore crystal structure.
To contribute to this debate, we have investigated several ordered and disordered configurations of stoichiometric La2Ce2O7 and La2Ce2O8. By investigating the heat of formation of the different systems, we found that under strongly oxidizing atmosphere (La2Ce2O8 stoichiometry) the disordered fluorite configuration was more stable than the pyrochlore or any of the other ordered structures. However, for the La2Ce2O7 stoichiometry, which did include oxygen vacancies, it was the pyrochlore configuration that turned out to be most stable. This shows that the amount of oxygen vacancies is a crucial aspect in this debate, allowing for the stability preference to move from the disordered fluorite to the pyrochlore structure. In addition, the calculated oxygen vacancy formation energies show a clear trend with regard to the amount of cerium in the tetrahedron surrounding the vacancy site (cf. Fig. 1a). More cerium results in a more stable structure, indicating that the formation of oxygen vacancies will provide a drive to more and more ordered structures, i.e. away from the random nature of the disordered fluorite structure. As before, it is clear that the introduction of oxygen vacancies has a significant influence on the properties of system.
Because XRD is often used indicate which of the two models is experimentally observed, we simulated XRD spectra for all structures studied. Interestingly we discovered that the pyrochlore structure shows the prototypical pyrochlore reflections, but they are extremely small (compared to what one would expect from typical pyrochlores such as La2Zr2O7) making them virtually impossible to observe in standard XRD measurements. Furthermore, we found that the other ordered structures show multiple peak splittings for the high intensity reflections, which would make them easily identifiable in experiments. As such, we show, using three different approaches, each pointing at the same result, that the ground state crystal structure for La2Ce2O7 is the pyrochlore structure.[1,3]
6. Atomic charges and charge transfer: Hirshfeld-I for solids
In this work, we also investigated the influence of doping on the internal charge distribution of the system. To have not only a qualitative, but also a quantitative understanding of the changes, we used the Hirshfeld-I atoms in molecules partitioning scheme. In implementing this partitioning scheme for periodic systems, we implemented one of the first atom based Hirshfeld-I partitioning schemes for solids. This implementation uses only grid based electron densities, making it independent of the solid state or quantum chemistry code used to generate it, or even the basis-set or potential type used in those codes.[4,5,6]
Using this program and the electron distributions obtained from the VASP solid state program we calculated the atomic charges for our set of group IV doped CeO2 systems.[7] Unlike all other properties for this set of systems, the atomic charges show the same behavior for both group IVa and IVb dopants. There is some charge transfer toward the dopant for the larger dopant elements, while there is some charge transfer away from the dopant for the smaller dopants. This charge transfer remains roughly limited to the first shell of oxygen atoms around the dopant, making this a very localized modification of the system.
7. Conclusion
The comparison of theoretical and experimental results is always a non-trivial exercise. Both have their own vocabulary, standards, methods, approximations and assumptions. Both also have their own limitations, which often remain partially or entirely invisible to their practitioners. This may sometimes give the impression that theoretical and experimental results for the same system are presented, where in reality the actual systems are wildly different, resulting in seemingly contradictory results. To be able to make a good comparison between theoretical results and experimental ones it is important to keep these differences in mind, and to know what the limitations are, but also to recognise the general aspects of the experiment/calculation.
In this work, we studied idealized systems using theoretical calculations and show that even in this case, or rather especially in this case, general trends are discovered. The resulting wealth of information has lead to insights both in the structure of a specific system such as lanthanum cerate (it allowed us to resolve some of the apparent contradictions in the experimental literature regarding the crystal structure of lanthanum cerate), and mechanical properties (such as the inverse relation between the bulk modulus and thermal expansion coefficient) of more general systems such as doped CeO2. It allowed us to predict new interesting candidates for doping CeO2, both as a bulk or surface dopant, but also to give a good starting guess for the required doping concentration for lattice matching of doped CeO2 with La2Zr2O7. Our implementation of the Hirshfeld-I partitioning scheme for solids opened up new possibilities for obtaining atomic charges in solids, and as a result allows for the quantitative study of charge transfer in solids, amongst other applications, such as input parameters for force field methods.
| <Ph.D. Thesis> | < Cover J. Comput. Chem. 34, issue 5 > |
|---|---|
 |
 |
Related Publications
-
Phys. Rev. B 84, 054110 (2011),
doi: 10.1103/PhysRevB.84.054110 -
Appl. Surf. Sci. 260, 32-35 (2012),
doi: 10.1016/j.apsusc.2012.01.032, -
J. Mater. Chem. 22, 8476 (2012),
doi:10.1039/C2JM15752G -
J. Comput. Chem. 34(5), 405-417 (2013),
doi:10.1002/jcc.23088 -
J. Comput. Chem. 34(5), 422-427 (2013),
doi:10.1002/jcc.23193 -
J. Comput. Chem. 34(5), i-ii (2013),
doi:10.1002/jcc.23239 -
J. Am. Ceram. Soc. 97(1), 258-266 (2014),
doi:10.1111/jace.12650 -
J. Mater. Chem. A 2, 13723-13737 (2014),
doi:10.1039/C4TA02449D