-
Filed under blog
-
July 9, 2026
We are excited to announce that Eleonora Thomas (GIMli project) has been selected as one of the finalists for the EUTalentOn 2026.
This September, in Brest, France, Eleonora is one of the 100 researchers who will work in multidisciplinary teams on challenges aligned with the five EU Mission Areas. Her materiomics background has inspired her to explore sustainability solutions, while her current PhD in the QuATOMs group continues to shape her scientific journey. Growing up in Curaçao has given her a deep appreciation for the ocean, and forms the core behind her motivation to protect our marine ecosystems. She is looking forward to collaborating with other motivated young researchers from across Europe, and contributing to the Restore Our Ocean and Waters mission!
The entire QuATOMs team is proud of her accomplishment, and will be rooting for her in September. Stay tunned for updates.
Permanent link to this article: https://dannyvanpoucke.be/et_talenton2026_1/
-
Filed under blog
-
May 27, 2026
On Wednesday May 27th, there was the annual conference of the Belgian Physical Society. As usual a good place to see what is happening in the belgian physics scene. Also the QuATOMs group was present, with posters on diamond growth (Emerick Guillaume), the GeV color center (Aylin Melan), and a talk on diamond color centers (myself).

BPS 2026 group picture.
Permanent link to this article: https://dannyvanpoucke.be/bps-conference-2026/
| Authors: |
Silviu Florin Acaru, Marc Comí, Panagiotis Falireas, Danny E. P. Vanpoucke, Richard Vendamme, and Katrien Bernaerts |
| Journal: |
Materials & Design 267, 116265 (2026) |
| doi: |
10.1016/j.matdes.2026.116265 |
| IF(2025): |
7.9 |
| export: |
bibtex |
| pdf: |
<Mat&Des_267> |
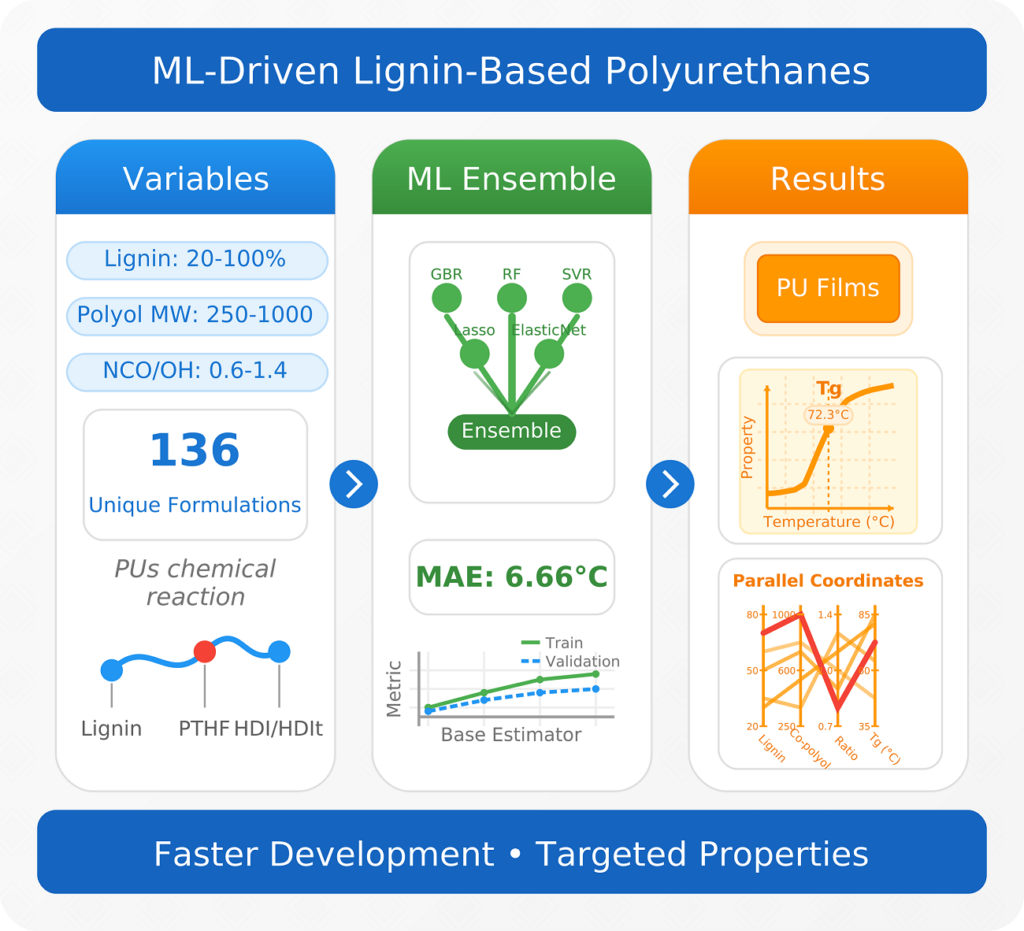 |
| Graphical Abstract: A machine learning ensemble accurately predicts glass transition temperatures for lignin-based polyurethanes within 6.66°C, despite being trained on a dataset of only 136 formulations. The model efficiently combines seven key features to overcome this data limitation. This optimized workflow generated over 4 million novel lignin-polyurethane formulations, with an intuitive interface accelerating the adoption of sustainable polyurethanes. |
Lignin-based polyurethanes (PUs) offer a compelling route towards sustainable material development, yet the challenge of designing chemical formulations with targeted properties, such as glass transition temperature (Tg), remains unresolved. In this work, we present a systematic approach, to explore key structural parameters—such as lignin content, polyol chain length, isocyanate functionality, and mixing ratios—across 136 unique formulations, creating a diverse dataset of ligninbased PUs. By harnessing this small dataset, we develop a machine learning (ML) ensemble model capable of accurately predicting Tg, with a mean absolute error of just 6.66°C on the validation set, surpassing the performance of conventional regression methods. Additionally, we enhance model interpretability by integrating advanced mapping techniques and employ an adaptive grid search algorithm to explore extrapolative scenarios. Our workflow, paired with a user-friendly interface, enables rapid discovery and optimization of formulations with desired properties. This study not only deepens the understanding of structure-property relationships in lignin-PUs but also provides a scalable ML-driven tool for designing sustainable materials with precision, highlighting the transformative potential of artificial intelligence in green chemistry and materials innovation.
Permanent link to this article: https://dannyvanpoucke.be/2026-paper_digiligninsylviu-en/
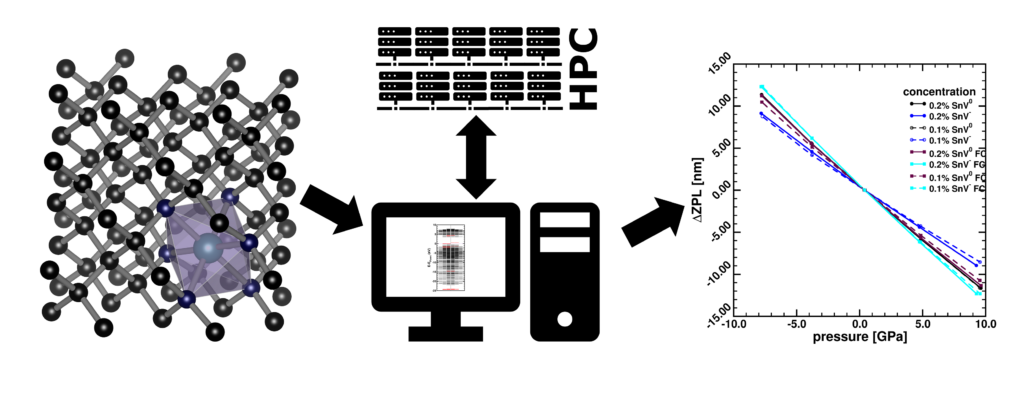 |
| Graphical Abstract: The SnV color center is modelled using first principle calculations to predict the zero-phonon-lines in diamond under hydrostatic strain. |
Among the group-IV vacancy color centers in diamond, the SnV holds promise for photonics based quantum applications. In this work, the Tin-Vacancy (SnV) zero-phonon line (ZPL) and its pressure coefficient are calculated using first principles approaches. The predicted absolute ZPL position is shown to be strongly influenced by the method and supercell size used. The results are therefore extrapolated to the dilute limit allowing for direct comparison with experiments. The importance of identifying the color-center related Kohn–Sham states is highlighted, as well as the shifting of these states due to electron excitations as well as supercell size and k-point position. In contrast to the absolute ZPL positions, the relative position of the SnV0 ZPL is consistently redshifted about 43 nm compared to the SnV– ZPL. In addition, the pressure coefficient is shown to be very robust over different methods, always resulting in a value of about 1.4 nm/GPa, for both SnV0 and SnV–. Finally, the computational accuracy and cost are put into perspective.
Permanent link to this article: https://dannyvanpoucke.be/2026-paper_snvcolorcenter/
It is a yearly habbit, the Hasselt diamond conference with the cryptic name SBDD. It stands for “Surface and Bulk Defects in Diamond”, though few remember as the accronym has been in common use for quite a while. This year, we celebrated the thirtied edition, or XXX using roman numerals. A celebratory edition which was filled with some special events, such as the XXX session (of course that was a fun group quize about the conference, what else did you think?), a caricaturist, a claw machine with SBDD goodies, a photobooth and a scientific poster/image competition. Indeed, the diamond community is a true scientific family when at SBDD.

SBDD XXX conference. Top left: Aylin Melan, Eleonora Thomas and Thijs van Wijk with the group poster. Top right: SBDD XXX poster prize winners. Bottom left: Caricature of Danny Vanpoucke on a beer coaster. Bottom right: Aylin with her prize winning poster.
QuATOMs was present with no less than 3 posters, and this year there was also good company from other theoretical contributors. We presented posters focussing on group-IV defects in diamond as well as our ambitions for the future. There was a huge number of posters (>170), with a lot of interest in modeling of defects. Any conference with poster sessions, also has a posterprize competition. This year, I’m happy to share that Aylin Melan won a Brillian poster prize at SBDD for her theoretical poster on GeV color centers, because of her skills at explaining the topic clearly for a broad (experimental) audience, as well as having a very nice poster. Congratulations Aylin!
Permanent link to this article: https://dannyvanpoucke.be/sbdd-xxx/
During the second semester of the academic year 2025-26 the QuATOMs group has the pleasure to welcome no less than 4 Bachelor intern students from Chemistry, and 2 junior master internships from biomedical sciences, in addition to our master materiomics student Bram Makowski, and Pedro Perrout who joined us from Brazil during the first semester. The coming few months, these students will be introduced into the marvellous world of computational research, and perform their first large true research project.
Welcome to all students of this year
- Bram Makowski (Materiomics), who is studying hybrid perovskites using molecular dynamics modelling.
- Pedro Perrout (extern: USP, Brazil), who studied the QM9 database, focussing on creating a chemical inspired fingerprint.
- Deborah Gagliardi (Chemistry, pre-materiomics), who will study H diffusion in diamond by means of molecular dynamics using veloxchem.
- Saba Heidarian (Chemistry, pre-materiomics), who will implement an LS-SVM classifier and study the QM9 dataset, placing her first steps into the world of machine learning.
- Brent Janssens (Chemistry), who will study vibrational spectra of diamond color centers and molecules using quantum chemical calculations .
- Aerts Milan (Chemistry), Jasper Schrijvers (Biomedical), and Lana Vandamme (Biomedical), who will datamine various datasets of molecules to learn more about the fundamental nature of chemical bonding.
Permanent link to this article: https://dannyvanpoucke.be/interships-of-2025-2026/
| Authors: |
Pieter Verding, Danny E.P. Vanpoucke, Yunus T. Aksoy, Tobias Corthouts, Maria R. Vetrano, and Wim Deferme |
| Journal: |
Adv. Mater. Technol. XX, YY (2025) |
| doi: |
10.1002/admt.202502104 |
| IF(2025): |
6.2 |
| export: |
bibtex |
| pdf: |
<AdvMaterTechnol_XX> |
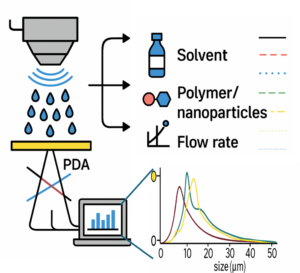 |
| Graphical Abstract: This study explores how machine learning models, trained on small experimental datasets obtained via Phase Doppler Anemometry (PDA), can accurately predict droplet size (D₃₂) in ultrasonic spray coating (USSC). By capturing the influence of ink complexity (solvent, polymer, nanoparticles), power, and flow rate, the model enables precise droplet control paving the way for optimized coatings in advanced functional materials. |
This study examines droplet formation in ultrasonic spray coating (USSC) as a function of ink formulation (solvent, polymer, nanoparticles). First, acetone with polyvinylidene fluoride (PVDF) at concentrations from 0-4.5 wt% is used to examine the effect of polymer additions. Additionally, acetone-based SiO2 nanofluids (0-10 g/L), are explored. Finally, the combination of both polymer (PVDF) and nanoparticles (SiO2) in acetone is studied. Droplet sizes are measured using Phase Doppler Anemometry under varying atomization power and flow rates. Machine Learning (ML) algorithms are employed to develop droplet size models from key spray parameters, including atomization power, flow rate, polymer concentration, and nanoparticle concentration. The model shows significantly higher accuracy than existing empirical models. The model is further validated on IPA-based inks with polyethylenimine (PEIE) or ZnO nanoparticles, and on acetone–cellulose acetate formulations, confirming its robustness across diverse ink systems. In addition to revealing the influence of coating parameters on the droplet formation and distribution, obtained both via experimental validation and ML, this study demonstrates that ML can be effectively applied to small experimental datasets, offering a robust framework for optimizing droplet formation and understanding key spray parameters in USSC for complex, unexplored inks enabling novel coating applications.
Permanent link to this article: https://dannyvanpoucke.be/2025-paper_mldroplets_pieterverding-en/
-
Filed under blog
-
December 13, 2025
| Authors: |
Rani Mary Joy, Miquel Cherta Garrido, Omar J.Y. Harb, Hendrik Jeuris, Rozita Rouzbahani, Jan D’Haen, Stephane Clemmen, Dries Van Thourhout, Danny E.P. Vanpoucke, Paulius Pobedinskas, and Ken Haenen |
| Journal: |
ACS Materials Lett. 8(1), 137-144 (2026) |
| doi: |
10.1021/acsmaterialslett.5c01218 |
| IF(2023): |
8.7 |
| export: |
bibtex |
| pdf: |
<ACSMaterialsLett_8> |
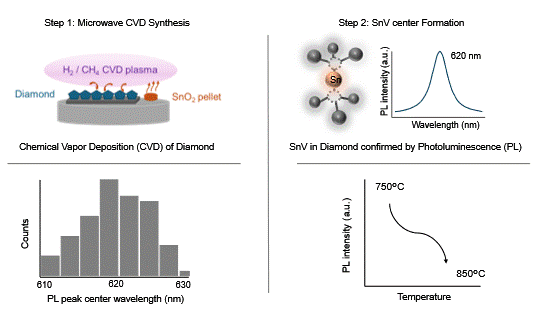 |
| Graphical Abstract: Experimental observation of SnV zero-phonon-lines in diamond. |
Group IV color centers in diamond are promising single-photon emitters for quantum information processing and networking. Among them, the tin-vacancy (SnV) center stands out due to its long spin coherence times at cryogenic temperatures above 1 K. While SnV centers have been realized using various fabrication routes, their in situ formation via microwave plasma-enhanced chemical vapor deposition (MW PE CVD) remains relatively unexplored. In this study, SnV centers, identified by a zero-phonon line (ZPL) near 620 nm, were synthesized in nanocrystalline diamond and free-standing microcrystalline diamond using tin oxide (SnO2) as a dopant source at substrate temperatures of 750°C and 850°C. Photoluminescence measurements reveal that lowering the substrate temperature enhances both the ZPL intensity and spatial uniformity of SnV centers. These results highlight substrate temperature as a key parameter for controlling SnV incorporation during MW PE CVD growth and provide insights into optimizing fabrication strategies for diamond-based quantum technologies.
Permanent link to this article: https://dannyvanpoucke.be/2025-paper_snvfabrication_ranimjoy-en/
| Authors: |
Goedele Roos, Danny E.P. Vanpoucke, Ralf Blossey, Marc F. Lensink, and Jane S. Murray |
| Journal: |
J. Chem. Phys. 163, 114112 (2025) |
| doi: |
10.1063/5.0268712 |
| IF(2023): |
3.1 |
| export: |
bibtex |
| pdf: |
<JChemPhys_163> |
 |
| Graphical Abstract: The Electrostatic Potential of water in different situations. On the left two interacting water molecules are shown, while on the right a water molecule interacting with a protein model representation is shown. |
The electrostatic potential plotted on varying contours (VS) of the electron density guides us in the
understanding of how water interactions exactly take place. Water—H2O—is extremely well balanced, having a hydrogen VS,max and an oxygen VS,min of similar magnitude. As such, it has the capacity to donate and accept hydrogen bonds equally well. This has implications for the interactions that water molecules form, which are reviewed here, first in water–small molecule models and then in complex sites as lactose and its crystals and in protein–protein interfaces. Favorable and unfavorable interactions are evaluated from the electrostatic potential plotted on varying contours of the electronic density, allowing these interactions to be readily visualized. As such, with one calculation, all interactions can be analyzed by gradually looking deeper into the electron density envelope and finding the nearly touching contour. Its relation with interaction strength has the electrostatic potential to be used in scoring functions. When properly implemented, we expect this approach to be valuable in modeling and structure validation, avoiding tedious interaction strength calculations. Here, applied to water interactions in a variety of systems, we conclude that all water interactions take the same general form, with water behaving as a “neutral” agent, allowing its interaction partner to determine if it donates or accepts a hydrogen bond, or both, as determined by the highest possible interaction strength(s).
Permanent link to this article: https://dannyvanpoucke.be/2025-paper-wateresp-roos-en/
-
Filed under blog
-
July 25, 2025
In light of the ever growing interest in AI and ML within the context of materials research, I’m guest editing a special issue together with Konstantin Klyukin from Auburn University. More information can be found on the flyer below.
(And yes the robot and diamond are AI generated, though it took some effort to get it to have the right number or arms, hold a diamond and look sideways at the same time. 😉

Permanent link to this article: https://dannyvanpoucke.be/special-issue-revolutions-in-the-integration-of-artificial-intelligence-and-machine-learning-in-carbon-based-materials-research/








