Category: 2024
| Authors: |
Emerick Y. Guillaume, Danny E. P. Vanpoucke, Rozita Rouzbahani, Luna Pratali Maffei, Matteo Pelucchi, Yoann Olivier, Luc Henrard, & Ken Haenen |
| Journal: |
Carbon 222, 118949 (2024) |
| doi: |
10.1016/j.carbon.2024.118949 |
| IF(2022): |
10.9 |
| export: |
bibtex |
| pdf: |
<Carbon> |
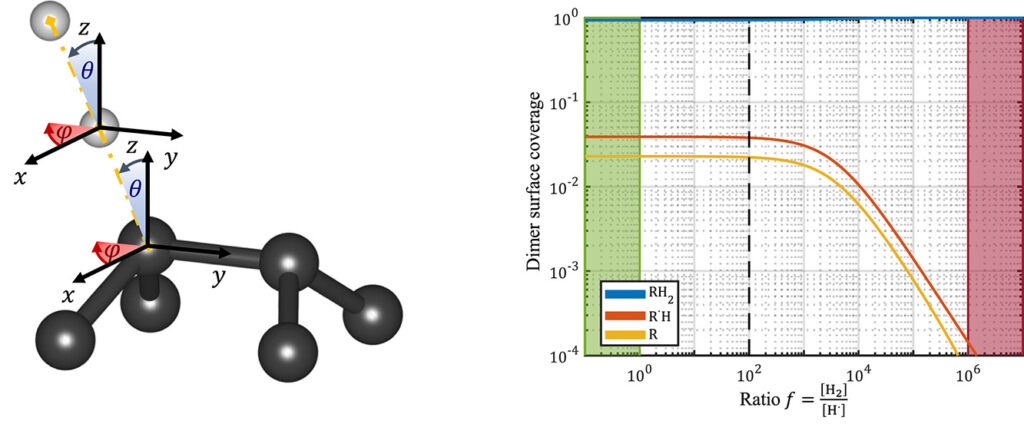 |
| Graphical Abstract: (left) Ball-and-stick representation of aH adsorption/desorption reaction mediated through a H radical. (right) Monte Carlo estimates of the H coverage of the diamond surface at different temperatures based on quantum mechanically determined reaction barriers and reaction rates. |
Hydrogen radical attacks and subsequent hydrogen migrations are considered to play an important role in the atomic-scale mechanisms of diamond chemical vapour deposition growth. We perform a comprehensive analysis of the reactions involving H-radical and vacancies on H-passivated diamond surfaces exposed to hydrogen radical-rich atmosphere. By means of first principles calculations—density functional theory and climbing image nudged elastic band method—transition states related to these mechanisms are identified and characterised. In addition, accurate reaction rates are computed using variational transition state theory. Together, these methods provide—for a broad range of temperatures and hydrogen radical concentrations—a picture of the relative likelihood of the migration or radical attack processes, along with a statistical description of the hydrogen coverage fraction of the (100) H-passivated surface, refining earlier results via a more thorough analysis of the processes at stake. Additionally, the migration of H-vacancy is shown to be anisotropic, and occurring preferentially across the dimer rows of the reconstructed surface. The approach used in this work can be generalised to other crystallographic orientations of diamond surfaces or other semiconductors.
Permanent link to this article: https://dannyvanpoucke.be/2024-paper-hadsorption-emerick-en/
| Authors: |
Emanuele Bosoni, Louis Beal, Marnik Bercx, Peter Blaha, Stefan Blügel, Jens Bröder, Martin Callsen, Stefaan Cottenier, Augustin Degomme, Vladimir Dikan, Kristjan Eimre, Espen Flage-Larsen, Marco Fornari, Alberto Garcia, Luigi Genovese, Matteo Giantomassi, Sebastiaan P. Huber, Henning Janssen, Georg Kastlunger, Matthias Krack, Georg Kresse, Thomas D. Kühne, Kurt Lejaeghere, Georg K. H. Madsen, Martijn Marsman, Nicola Marzari, Gregor Michalicek, Hossein Mirhosseini, Tiziano M. A. Müller, Guido Petretto, Chris J. Pickard, Samuel Poncé, Gian-Marco Rignanese, Oleg Rubel, Thomas Ruh, Michael Sluydts, Danny E.P. Vanpoucke, Sudarshan Vijay, Michael Wolloch, Daniel Wortmann, Aliaksandr V. Yakutovich, Jusong Yu, Austin Zadoks, Bonan Zhu, and Giovanni Pizzi |
| Journal: |
Nature Reviews Physics 6(1), (2024) |
| doi: |
web only |
| IF(2021): |
36.273 |
| export: |
NA |
| pdf: |
<NatRevPhys> |
The cover of this issue shows an artistic representation of the equations of state of the periodic table elements, calculated using two all-electron codes in each of the 10 crystal structure configurations shown on the table. The cover image is based on the Perspective Article How to verify the precision of density-functional-theory implementations via reproducible and universal workflows by E. Bosoni et al., https://doi.org/10.1038/s42254-023-00655-3. (The related paper can be found here.)

Cover Nature Reviews Physics: Accuracy of DFT modeling in solids
Permanent link to this article: https://dannyvanpoucke.be/paper2024_accuracycover-en/
| Authors: |
Emanuele Bosoni, Louis Beal, Marnik Bercx, Peter Blaha, Stefan Blügel, Jens Bröder, Martin Callsen, Stefaan Cottenier, Augustin Degomme, Vladimir Dikan, Kristjan Eimre, Espen Flage-Larsen, Marco Fornari, Alberto Garcia, Luigi Genovese, Matteo Giantomassi, Sebastiaan P. Huber, Henning Janssen, Georg Kastlunger, Matthias Krack, Georg Kresse, Thomas D. Kühne, Kurt Lejaeghere, Georg K. H. Madsen, Martijn Marsman, Nicola Marzari, Gregor Michalicek, Hossein Mirhosseini, Tiziano M. A. Müller, Guido Petretto, Chris J. Pickard, Samuel Poncé, Gian-Marco Rignanese, Oleg Rubel, Thomas Ruh, Michael Sluydts, Danny E.P. Vanpoucke, Sudarshan Vijay, Michael Wolloch, Daniel Wortmann, Aliaksandr V. Yakutovich, Jusong Yu, Austin Zadoks, Bonan Zhu, and Giovanni Pizzi |
| Journal: |
Nature Reviews Physics 6(1), 45-58 (2024) |
| doi: |
10.1038/s42254-023-00655-3 |
| IF(2021): |
36.273 |
| export: |
bibtex |
| pdf: |
<NatRevPhys>
<ArXiv:2305.17274> |
 |
| Graphical Abstract: “We hope our dataset will be a reference for the field for years to come,” says Giovanni Pizzi, leader of the Materials Software and Data Group at the Paul Scherrer Institute PSI, who led the study. (Image: Paul Scherrer Insitute / Giovanni Pizzi) |
Density-functional theory methods and codes adopting periodic boundary conditions are extensively used in condensed matter physics and materials science research. In 2016, their precision (how well properties computed with different codes agree among each other) was systematically assessed on elemental crystals: a first crucial step to evaluate the reliability of such computations. In this Expert Recommendation, we discuss recommendations for verification studies aiming at further testing precision and transferability of density-functional-theory computational approaches and codes. We illustrate such recommendations using a greatly expanded protocol covering the whole periodic table from Z = 1 to 96 and characterizing 10 prototypical cubic compounds for each element: four unaries and six oxides, spanning a wide range of coordination numbers and oxidation states. The primary outcome is a reference dataset of 960 equations of state cross-checked between two all-electron codes, then used to verify and improve nine pseudopotential-based approaches. Finally, we discuss the extent to which the current results for total energies can be reused for different goals.
Permanent link to this article: https://dannyvanpoucke.be/paper-aiidaconsortium2023-en/



