Happy 2025
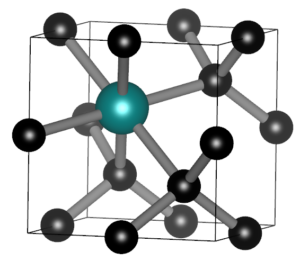
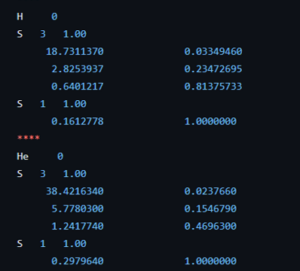
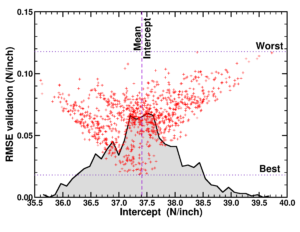
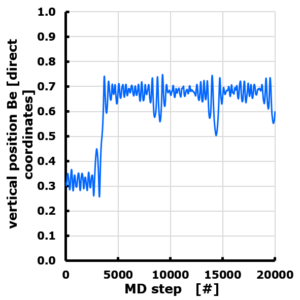
2023 and 2024 have been an intense ride, with both the materiomics program, the tenure track and the research group. Since the previous overview in 2022, the QuATOMs group has seen some growth with the arrival of three new members (Pauline Castenetto, Thijs van Wijk, and Aylin Melan). In addition, Emerick Guillaume defended his PhD on the study of diamond growth.
These are not the only things which happened the last two years, so let us look back at 2023 and 2024 one last time, keeping up with tradition.
1. Publications: +4 (and currently a handful in progress)
-
J. Alloys Compd. 936, 168138 (2023),
doi: 10.1016/j.jallcom.2022.168138 {IF(2021)=6.371} -
J. Mol. Liq. 384, 122172 (2023),
doi: 10.1016/j.molliq.2023.122172 {IF(2021)=6.633} -
Nat. Rev. Phys. 6(1), 45-58 (2024),
doi: 10.1038/s42254-023-00655-3 {IF(2021)=36.273} -
Carbon 222, 118949 (2024),
doi: 10.1016/j.carbon.2024.118949 {IF(2022)=10.9}
2. Cover publication: +1

Cover Nature Reviews Physics: Accuracy of DFT
-
Nat. Rev. Phys. 6(1), (2024),
doi: na (web) {IF(2021)=36.273}
3. Project proposals accepted: +2
- QuantumLignin: Elucidating the interactions of lignin building blocks with their environment for the creation of additive models by means of quantum mechanical modelling.
- AI-accelerated quantum mechanical modelling of the optical properties of semiconductor materials: from colour centres in diamond to transition-metal oxides.
4. Completed refereeing tasks: +20
- ACS Photon
- Journal of Physics D: Applied Physics (2x)
- Philosophical Transactions (2x)
- Journal of Materials Chemistry A (2x)
- Journal of Physical Chemistry (3x)
- Diamond and Related Materials (11x)
5. Conferences & seminars: +7/+1 (Attended & Organised)
- SBDD XXVII & SBDD XXVIII, Hasselt University, Belgium, March 15th-17th, 2023 & February 28th-March 1st, 2024 [poster presentations, PhD students & MSc student]
- EAB-workshop on AI in Higher Education, UHasselt, Belgium, April 24th, 2024 [oral presentation]
- DFT-2024: 20th International Conference on Density Functional Theory and its Applications, Paris, France, August 25th-30th, 2024 [oral presentation]
- E-MRS Spring meeting 2023 & 2024, Strassbourg, France, May 29th-June 2nd, 2023 & May 27th-31st, 2024 [oral presentations]
- Summer School: “Materiomics: Innovative Materials From Healthcare Across Quantum To Sustainable Technologies”, UHasselt, Belgium, September 4th-6th, 2024 [member of Organizating Committee; seminar]
- NISM seminar: “Extreme Machine Learning – When the average model knows best” (Prof. L. Henrard), UNamur, Belgium, September 12th, 2024 [invited seminar]
6. Supervised students:
- BSc Projects Physics: 4
- MSc Projects Materiomics: 3
- Internships: 2
7. Hive-STM program:
- 63 new users in 2023 & 2024 (making for a total of 587 users)