Tag: DFT
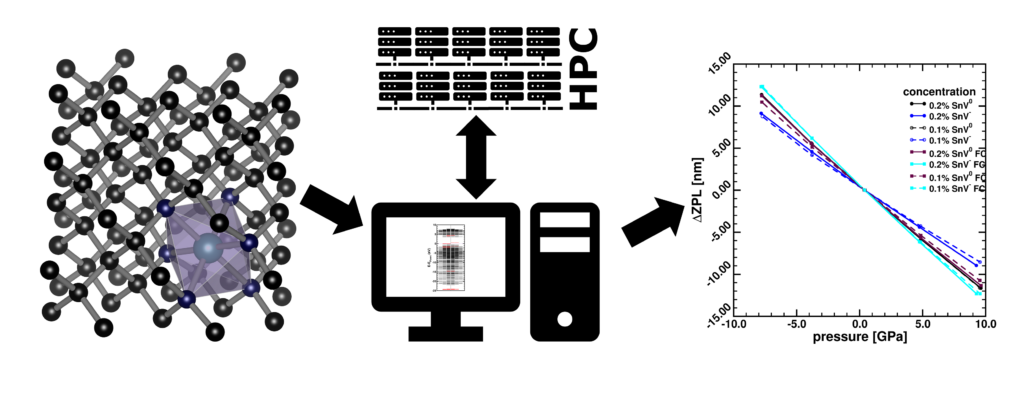 |
| Graphical Abstract: The SnV color center is modelled using first principle calculations to predict the zero-phonon-lines in diamond under hydrostatic strain. |
Among the group-IV vacancy color centers in diamond, the SnV holds promise for photonics based quantum applications. In this work, the Tin-Vacancy (SnV) zero-phonon line (ZPL) and its pressure coefficient are calculated using first principles approaches. The predicted absolute ZPL position is shown to be strongly influenced by the method and supercell size used. The results are therefore extrapolated to the dilute limit allowing for direct comparison with experiments. The importance of identifying the color-center related Kohn–Sham states is highlighted, as well as the shifting of these states due to electron excitations as well as supercell size and k-point position. In contrast to the absolute ZPL positions, the relative position of the SnV0 ZPL is consistently redshifted about 43 nm compared to the SnV– ZPL. In addition, the pressure coefficient is shown to be very robust over different methods, always resulting in a value of about 1.4 nm/GPa, for both SnV0 and SnV–. Finally, the computational accuracy and cost are put into perspective.
Permanent link to this article: https://dannyvanpoucke.be/2026-paper_snvcolorcenter/
| Authors: |
Goedele Roos, Danny E.P. Vanpoucke, Ralf Blossey, Marc F. Lensink, and Jane S. Murray |
| Journal: |
J. Chem. Phys. 163, 114112 (2025) |
| doi: |
10.1063/5.0268712 |
| IF(2023): |
3.1 |
| export: |
bibtex |
| pdf: |
<JChemPhys_163> |
 |
| Graphical Abstract: The Electrostatic Potential of water in different situations. On the left two interacting water molecules are shown, while on the right a water molecule interacting with a protein model representation is shown. |
The electrostatic potential plotted on varying contours (VS) of the electron density guides us in the
understanding of how water interactions exactly take place. Water—H2O—is extremely well balanced, having a hydrogen VS,max and an oxygen VS,min of similar magnitude. As such, it has the capacity to donate and accept hydrogen bonds equally well. This has implications for the interactions that water molecules form, which are reviewed here, first in water–small molecule models and then in complex sites as lactose and its crystals and in protein–protein interfaces. Favorable and unfavorable interactions are evaluated from the electrostatic potential plotted on varying contours of the electronic density, allowing these interactions to be readily visualized. As such, with one calculation, all interactions can be analyzed by gradually looking deeper into the electron density envelope and finding the nearly touching contour. Its relation with interaction strength has the electrostatic potential to be used in scoring functions. When properly implemented, we expect this approach to be valuable in modeling and structure validation, avoiding tedious interaction strength calculations. Here, applied to water interactions in a variety of systems, we conclude that all water interactions take the same general form, with water behaving as a “neutral” agent, allowing its interaction partner to determine if it donates or accepts a hydrogen bond, or both, as determined by the highest possible interaction strength(s).
Permanent link to this article: https://dannyvanpoucke.be/2025-paper-wateresp-roos-en/
For the second year in a row, we are organising a summerschool with the master materiomics program, oriented at students in their second or third bachelor chemistry or physics. Within this summerschool the students are introduced into the various topics which play an important role in materials research. Today, as part of the quantum pillar, I had the pleasure to introduce the students into the world of computational research, with a focus on the application for quantum mechanical modelling. We learned for example, that it is practically impossible to store the wavefunction of a simple small molecule like benzene, it would require more great deal more than a mole of galaxies in mass to store it. With Density Functional Theory on the other hand, you can easily investigate it on a modern day laptop, as you only need the electron density.

Materiomics summerschool of 2025.
Permanent link to this article: https://dannyvanpoucke.be/summerschool-materiomics/
| Authors: |
Thijs G.I. van Wijk, E. Aylin Melan, Rani Mary Joy, Emerick Y. Guillaume, Paulius Pobedinskas, Ken Haenen, and Danny E.P. Vanpoucke |
| Journal: |
Carbon 234, 119928 (2025) |
| doi: |
10.1016/j.carbon.2024.119928 |
| IF(2024): |
10.5 |
| export: |
bibtex |
| pdf: |
<Carbon> |
 |
| Graphical Abstract: Schematic representation of the impact of hydrostatic and linear strain on the Zero Phonon Line of the neutral GeV defect in diamond. |
Color centers in diamond, such as the GeV center, are promising candidates for quantum-based applications. Here, we investigate the impact of strain on the zero-phonon line (ZPL) position of GeV0. Both hydrostatic and linear strain are modeled using density functional theory for GeV0concentrations of 1.61 % down to 0.10 %. We present qualitative and quantitative differences between the two strain types: for hydrostatic tensile and compressive strain, red- and blue-shifted ZPL positions are expected, respectively, with a linear relation between the ZPL shift and the experienced stress. By calculating the ZPL shift for varying GeV0 concentrations, a shift of 0.15 nm/GPa (0.38 meV/GPa) is obtained at experimentally relevant concentrations using a hybrid functional. In contrast, only red-shifted ZPL are found for tensile and compressive linear strain along the ⟨100⟩ direction. The calculated ZPL shift exceeds that of hydrostatic strain by at least one order of magnitude, with a significant difference between tensile and compressive strains: 3.2 and 4.8 nm/GPa (8.1 and 11.7 meV/GPa), respectively. In addition, a peak broadening is expected
due to the lifted degeneracy of the GeV0 eg state, calculated to be about 6 meV/GPa. These calculated results are placed in perspective with experimental observations, showing values of ZPL shifts and splittings of comparable magnitude.
Permanent link to this article: https://dannyvanpoucke.be/2025-paper-strainedgev-en/
 Since the first of January 2025, the QuATOMs group has been strengthened with a new member: Minh-Thu Bui.
Since the first of January 2025, the QuATOMs group has been strengthened with a new member: Minh-Thu Bui.
She is an expert in polymer chemistry, with a MSc in Polymers for advanced Technologies from the university of Grénoble. The coming four years she will be working on the QuantumLignin project. In this project, she’ll investigate the structure-property relations of lignin building blocks, with the aim of creating an additive model suitable for predicting the properties of mixed lignin samples. With her life motto: “Don’t wait for the perfect moment. Take the moment and make it perfect.” I’m sure we can expect great things to happen in the theoretical lignin field, the coming years.
Welcome to the QuATOMs team, we look forward to your enthusiasm and the intuition you bring to the team.
Permanent link to this article: https://dannyvanpoucke.be/new-quatoms-group-member-minh-thu-bui/
| Authors: |
Asif Iqbal Bhatti, Sandeep Kumar, Catharina Jaeken, Michael Sluydts, Danny E.P. Vanpoucke, and Stefaan Cottenier |
| Journal: |
Journal of Materials Chemistry A 13, 526-539 (2025) |
| doi: |
10.1039/D4TA06603K |
| IF(2024): |
10.7 |
| export: |
bibtex |
| pdf: |
<J.Mat.Chem.A> |
 |
| Graphical Abstract: Schematic representation of the LPS material and the variation of results obtained due to slight changes in settings within a High Throughput workflow. |
High-throughput computational screening has become a powerful tool in materials science for identifying promising candidates for specific applications. However, the effectiveness of these methods relies heavily on the accuracy and appropriateness of the underlying models and assumptions. In this study, we use the popular argyrodite solid-state electrolyte family Li6PS5X (X = Cl, Br, I) as a case study to critically examine key steps in high-throughput workflows and highlight potential pitfalls. We demonstrate some of these pitfalls by highlighting the importance of careful structural considerations, including symmetry breaking and site disorder, and examine the difference between 0 K thermodynamic stability and finite-temperature stability based on temperature-dependent Gibbs free energy calculations. Furthermore, we explore the implications of these findings for the ranking of candidate materials in a mini-throughput study in a search space of isovalent analogs to Li6PS5Cl. As a result of these findings, our work underscores the need for balanced trade-offs between computational efficiency and accuracy in high-throughput screenings, and offers guidance for designing more robust workflows that can better bridge the gap between computational predictions and experimental realities.
Permanent link to this article: https://dannyvanpoucke.be/2025-paper-thedevilinthedetails-en/
 During the last year, Esin Aylin Melan worked hard at her MSc Thesis within the QuATOMs group. Her research focus was centered on the impact of strain on the zero-phonon-line of the GeV color center in diamond. This work she presented, together with Thijs van Wijk, at the SBDD conference in Hasselt, and was presented as well at both the BPS and EMRS spring meeting of 2024. Before the summer she gave her (very good and enthusiastic) final presentation of the MSc thesis results, bringing her first real research project to good end. (Paper will follow later 🙂 )
During the last year, Esin Aylin Melan worked hard at her MSc Thesis within the QuATOMs group. Her research focus was centered on the impact of strain on the zero-phonon-line of the GeV color center in diamond. This work she presented, together with Thijs van Wijk, at the SBDD conference in Hasselt, and was presented as well at both the BPS and EMRS spring meeting of 2024. Before the summer she gave her (very good and enthusiastic) final presentation of the MSc thesis results, bringing her first real research project to good end. (Paper will follow later 🙂 )
Recently, she also received the great news that she was awarded a bilateral PhD Scholarship between UHasselt & UNamur. So from September first, she has started working on the modeling of color centers in diamond and oxides for the coming four years. Welcome to the QuATOMs team, and congratulations on the scholarship. We look forward to the enthusiasm and insights you’ll bring to the team.
Permanent link to this article: https://dannyvanpoucke.be/new-quatoms-group-member/
| Authors: |
Emerick Y. Guillaume, Danny E. P. Vanpoucke, Rozita Rouzbahani, Luna Pratali Maffei, Matteo Pelucchi, Yoann Olivier, Luc Henrard, & Ken Haenen |
| Journal: |
Carbon 222, 118949 (2024) |
| doi: |
10.1016/j.carbon.2024.118949 |
| IF(2022): |
10.9 |
| export: |
bibtex |
| pdf: |
<Carbon> |
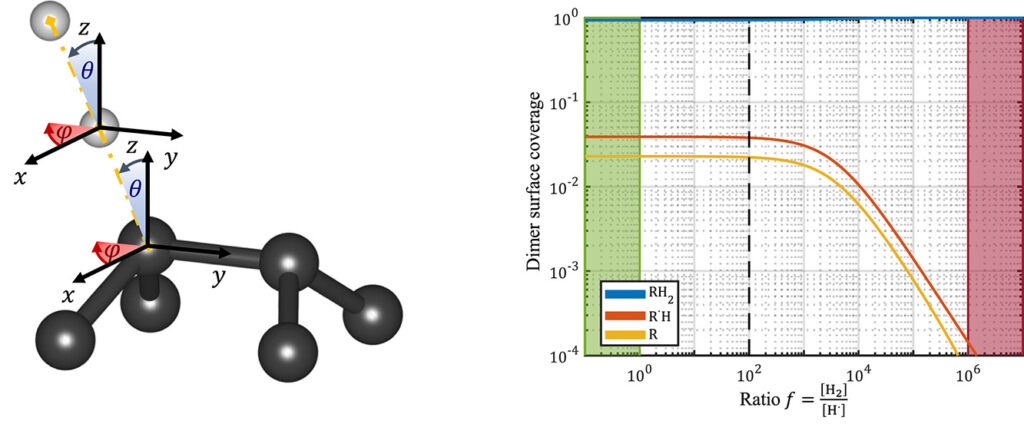 |
| Graphical Abstract: (left) Ball-and-stick representation of aH adsorption/desorption reaction mediated through a H radical. (right) Monte Carlo estimates of the H coverage of the diamond surface at different temperatures based on quantum mechanically determined reaction barriers and reaction rates. |
Hydrogen radical attacks and subsequent hydrogen migrations are considered to play an important role in the atomic-scale mechanisms of diamond chemical vapour deposition growth. We perform a comprehensive analysis of the reactions involving H-radical and vacancies on H-passivated diamond surfaces exposed to hydrogen radical-rich atmosphere. By means of first principles calculations—density functional theory and climbing image nudged elastic band method—transition states related to these mechanisms are identified and characterised. In addition, accurate reaction rates are computed using variational transition state theory. Together, these methods provide—for a broad range of temperatures and hydrogen radical concentrations—a picture of the relative likelihood of the migration or radical attack processes, along with a statistical description of the hydrogen coverage fraction of the (100) H-passivated surface, refining earlier results via a more thorough analysis of the processes at stake. Additionally, the migration of H-vacancy is shown to be anisotropic, and occurring preferentially across the dimer rows of the reconstructed surface. The approach used in this work can be generalised to other crystallographic orientations of diamond surfaces or other semiconductors.
Permanent link to this article: https://dannyvanpoucke.be/2024-paper-hadsorption-emerick-en/
| Authors: |
Emanuele Bosoni, Louis Beal, Marnik Bercx, Peter Blaha, Stefan Blügel, Jens Bröder, Martin Callsen, Stefaan Cottenier, Augustin Degomme, Vladimir Dikan, Kristjan Eimre, Espen Flage-Larsen, Marco Fornari, Alberto Garcia, Luigi Genovese, Matteo Giantomassi, Sebastiaan P. Huber, Henning Janssen, Georg Kastlunger, Matthias Krack, Georg Kresse, Thomas D. Kühne, Kurt Lejaeghere, Georg K. H. Madsen, Martijn Marsman, Nicola Marzari, Gregor Michalicek, Hossein Mirhosseini, Tiziano M. A. Müller, Guido Petretto, Chris J. Pickard, Samuel Poncé, Gian-Marco Rignanese, Oleg Rubel, Thomas Ruh, Michael Sluydts, Danny E.P. Vanpoucke, Sudarshan Vijay, Michael Wolloch, Daniel Wortmann, Aliaksandr V. Yakutovich, Jusong Yu, Austin Zadoks, Bonan Zhu, and Giovanni Pizzi |
| Journal: |
Nature Reviews Physics 6(1), (2024) |
| doi: |
web only |
| IF(2021): |
36.273 |
| export: |
NA |
| pdf: |
<NatRevPhys> |
The cover of this issue shows an artistic representation of the equations of state of the periodic table elements, calculated using two all-electron codes in each of the 10 crystal structure configurations shown on the table. The cover image is based on the Perspective Article How to verify the precision of density-functional-theory implementations via reproducible and universal workflows by E. Bosoni et al., https://doi.org/10.1038/s42254-023-00655-3. (The related paper can be found here.)

Cover Nature Reviews Physics: Accuracy of DFT modeling in solids
Permanent link to this article: https://dannyvanpoucke.be/paper2024_accuracycover-en/
| Authors: |
Emanuele Bosoni, Louis Beal, Marnik Bercx, Peter Blaha, Stefan Blügel, Jens Bröder, Martin Callsen, Stefaan Cottenier, Augustin Degomme, Vladimir Dikan, Kristjan Eimre, Espen Flage-Larsen, Marco Fornari, Alberto Garcia, Luigi Genovese, Matteo Giantomassi, Sebastiaan P. Huber, Henning Janssen, Georg Kastlunger, Matthias Krack, Georg Kresse, Thomas D. Kühne, Kurt Lejaeghere, Georg K. H. Madsen, Martijn Marsman, Nicola Marzari, Gregor Michalicek, Hossein Mirhosseini, Tiziano M. A. Müller, Guido Petretto, Chris J. Pickard, Samuel Poncé, Gian-Marco Rignanese, Oleg Rubel, Thomas Ruh, Michael Sluydts, Danny E.P. Vanpoucke, Sudarshan Vijay, Michael Wolloch, Daniel Wortmann, Aliaksandr V. Yakutovich, Jusong Yu, Austin Zadoks, Bonan Zhu, and Giovanni Pizzi |
| Journal: |
Nature Reviews Physics 6(1), 45-58 (2024) |
| doi: |
10.1038/s42254-023-00655-3 |
| IF(2021): |
36.273 |
| export: |
bibtex |
| pdf: |
<NatRevPhys>
<ArXiv:2305.17274> |
 |
| Graphical Abstract: “We hope our dataset will be a reference for the field for years to come,” says Giovanni Pizzi, leader of the Materials Software and Data Group at the Paul Scherrer Institute PSI, who led the study. (Image: Paul Scherrer Insitute / Giovanni Pizzi) |
Density-functional theory methods and codes adopting periodic boundary conditions are extensively used in condensed matter physics and materials science research. In 2016, their precision (how well properties computed with different codes agree among each other) was systematically assessed on elemental crystals: a first crucial step to evaluate the reliability of such computations. In this Expert Recommendation, we discuss recommendations for verification studies aiming at further testing precision and transferability of density-functional-theory computational approaches and codes. We illustrate such recommendations using a greatly expanded protocol covering the whole periodic table from Z = 1 to 96 and characterizing 10 prototypical cubic compounds for each element: four unaries and six oxides, spanning a wide range of coordination numbers and oxidation states. The primary outcome is a reference dataset of 960 equations of state cross-checked between two all-electron codes, then used to verify and improve nine pseudopotential-based approaches. Finally, we discuss the extent to which the current results for total energies can be reused for different goals.
Permanent link to this article: https://dannyvanpoucke.be/paper-aiidaconsortium2023-en/





 Since the first of January 2025, the
Since the first of January 2025, the 
 During the last year,
During the last year, 
