Category: 2020
| Authors: |
Danny E. P. Vanpoucke, Onno S. J. van Knippenberg, Ko Hermans, Katrien V. Bernaerts, and Siamak Mehrkanoon |
| Journal: |
Journal of Applied Physics 128, 054901 (2020) |
| doi: |
10.1063/5.0012285 |
| IF(2019): |
2.286 |
| export: |
bibtex |
| pdf: |
<JApplPhys> (Open Access) |
| github: |
<Amadeus> |
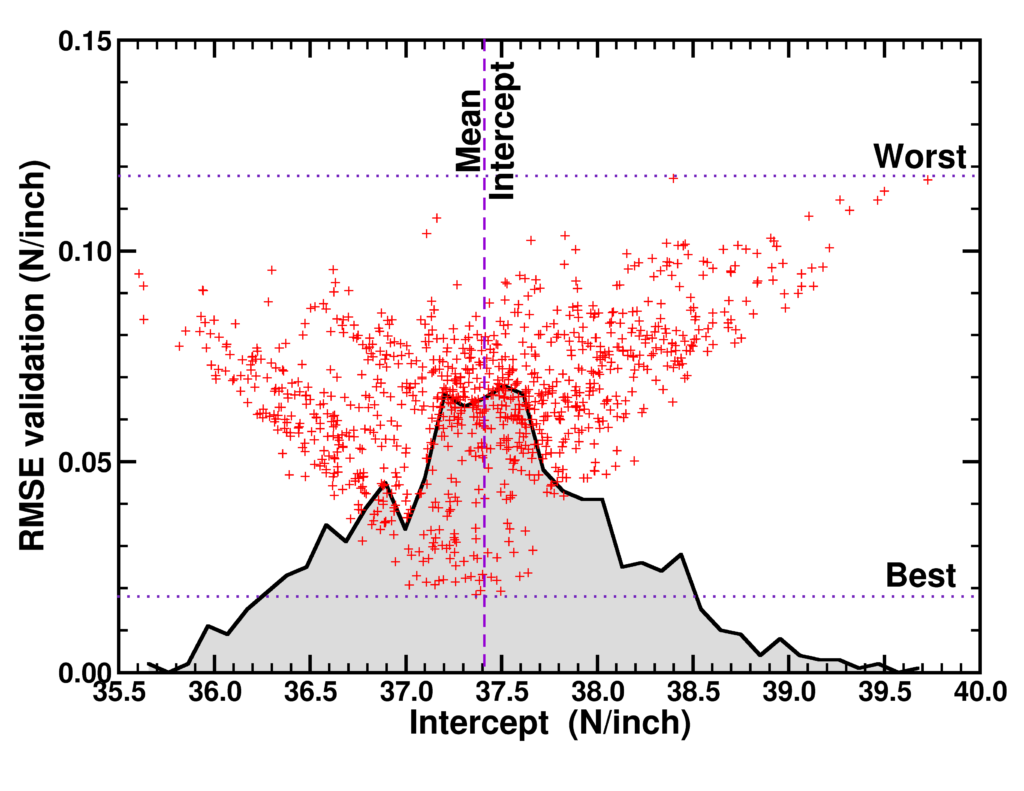 |
| Graphical Abstract: Correlation plot of the RMSE of the validation set and the intercept value for linear model instances trained on 1000 subsets of a 25 point data set. The distribution of the correlation data is indicated by the black curve. |
Machine Learning is quickly becoming an important tool in modern materials design. Where many of its successes are rooted in huge data sets, the most common applications in academic and industrial materials design deal with data sets of at best a few tens of data points. Harnessing the power of Machine Learning in this context is therefore of considerable importance. In this work, we investigate the intricacies introduced by these small data sets. We show that individual data points introduce a significant chance factor in both model training and quality measurement. This chance factor can be mitigated by the introduction of an ensemble-averaged model. This model presents the highest accuracy while at the same time it is robust with regard to changing data set size. Furthermore, as only a single model instance needs to be stored and evaluated, it provides a highly efficient model for prediction purposes, ideally suited for the practical materials scientist.
Permanent link to this article: https://dannyvanpoucke.be/paper_averagemodel-en/
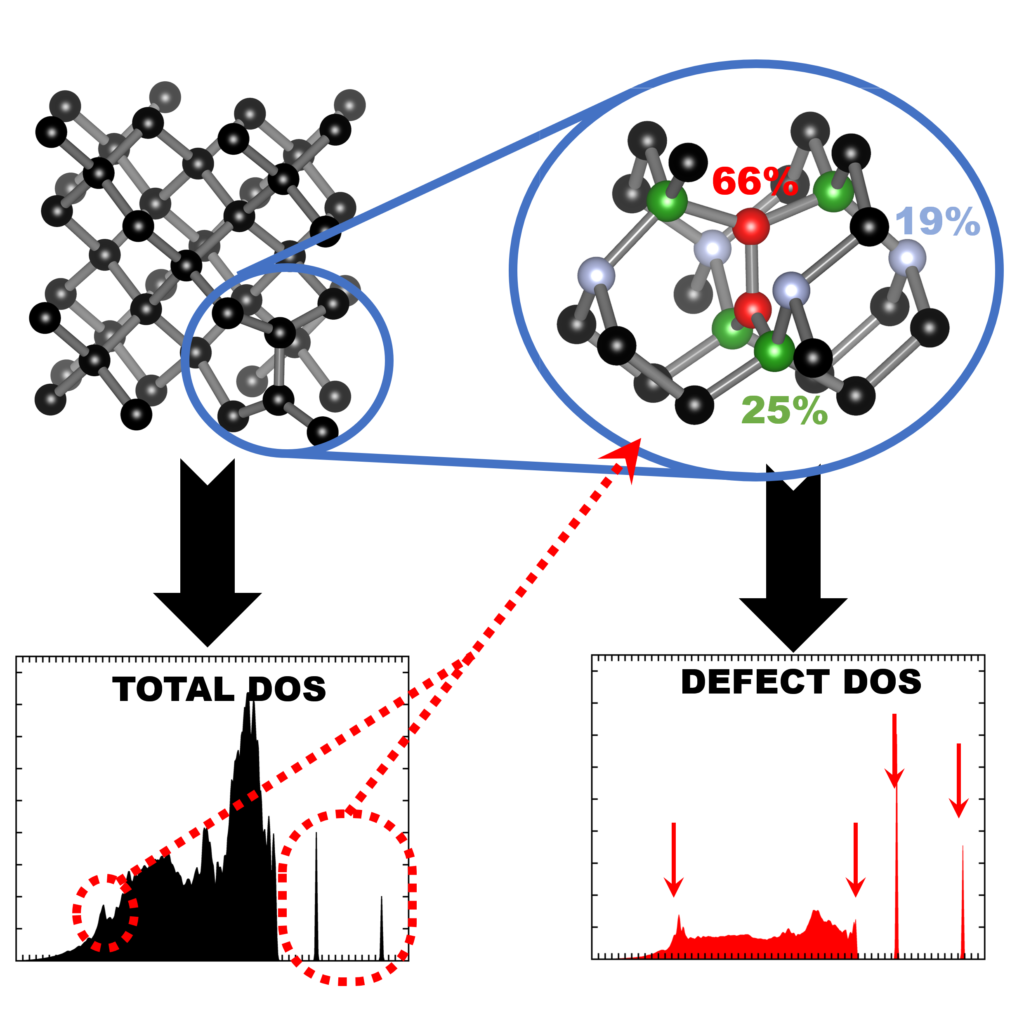 |
| Graphical Abstract: Finger printing defects in diamond through the creation of the vibrational spectrum of a defect. |
Vibrational spectroscopy techniques are some of the most-used tools for materials
characterization. Their simulation is therefore of significant interest, but commonly
performed using low cost approximate computational methods, such as force-fields.
Highly accurate quantum-mechanical methods, on the other hand are generally only used
in the context of molecules or small unit cell solids. For extended solid systems,
such as defects, the computational cost of plane wave based quantum mechanical simulations
remains prohibitive for routine calculations. In this work, we present a computational scheme
for isolating the vibrational spectrum of a defect in a solid. By quantifying the defect character
of the atom-projected vibrational spectra, the contributing atoms are identified and the strength
of their contribution determined. This method could be used to systematically improve phonon
fragment calculations. More interestingly, using the atom-projected vibrational spectra of the
defect atoms directly, it is possible to obtain a well-converged defect spectrum at lower
computational cost, which also incorporates the host-lattice interactions. Using diamond as
the host material, four point-defect test cases, each presenting a distinctly different
vibrational behaviour, are considered: a heavy substitutional dopant (Eu), two intrinsic
point-defects (neutral vacancy and split interstitial), and the negatively charged N-vacancy
center. The heavy dopant and split interstitial present localized modes at low and high
frequencies, respectively, showing little overlap with the host spectrum. In contrast, the
neutral vacancy and the N-vacancy center show a broad contribution to the upper spectral range
of the host spectrum, making them challenging to extract. Independent of the vibrational behaviour,
the main atoms contributing to the defect spectrum can be clearly identified. Recombination of
their atom-projected spectra results in the isolated spectrum of the point-defect.
Permanent link to this article: https://dannyvanpoucke.be/paper_vibrdefect-en/
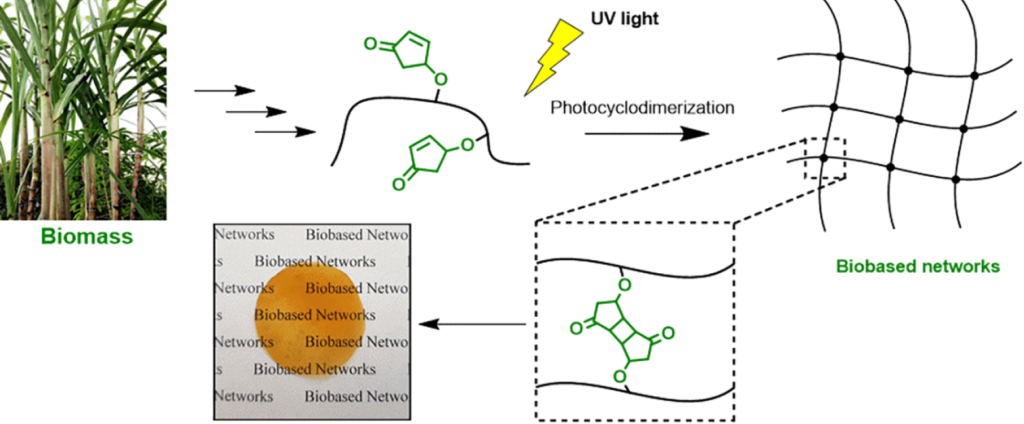 |
| Graphical Abstract: The formation of biobased polyacrylates. |
The controlled polymerization of a new biobased monomer, 4-oxocyclopent-2-en-1-yl acrylate (4CPA), was
established via reversible addition−fragmentation chain transfer (RAFT) (co)polymerization to yield polymers bearing pendent cyclopentenone units. 4CPA contains two reactive functionalities, namely, a vinyl group and an internal double bond, and is an unsymmetrical monomer. Therefore, competition between the internal double bond and the vinyl group eventually leads to gel formation. With RAFT polymerization, when aiming for a degree of polymerization (DP) of 100, maximum 4CPA conversions of the vinyl group between 19.0 and 45.2% were obtained without gel formation or extensive broadening of the dispersity. When the same conditions were applied in the copolymerization of 4CPA with lauryl acrylate (LA), methyl acrylate (MA), and isobornyl acrylate, 4CPA conversions of the vinyl group between 63 and 95% were reached. The additional functionality of 4CPA in copolymers was demonstrated by model studies with 4-oxocyclopent-2-en-1-yl acetate (1), which readily dimerized under UV light via [2 + 2] photocyclodimerization. First-principles quantum mechanical simulations supported the experimental observations made in NMR. Based on the calculated energetics and chemical shifts, a mixture of head-to-head and head-to-tail dimers of (1) were identified. Using the dimerization mechanism, solvent-cast LA and MA copolymers containing 30 mol % 4CPA were cross-linked under UV light to obtain thin films. The cross-linked films were characterized by dynamic scanning calorimetry, dynamic mechanical analysis, IR, and swelling experiments. This is the first case where 4CPA is described as a monomer for functional biobased polymers that can undergo additional UV curing via photodimerization.
Permanent link to this article: https://dannyvanpoucke.be/paper_nmrjules-en/
| Authors: |
Viraj Damle, Kaiqi Wu, Oreste De Luca, Natalia Ortí-Casañ, Neda Norouzi, Aryan Morita, Joop de Vries, Hans Kaper, Inge Zuhorn, Ulrich Eisel, Danny E.P. Vanpoucke, Petra Rudolf, and Romana Schirhagl, |
| Journal: |
Carbon 162, 1-12 (2020) |
| doi: |
10.1016/j.carbon.2020.01.115 |
| IF(2019): |
8.821 |
| export: |
bibtex |
| pdf: |
<Carbon> (Open Access) |
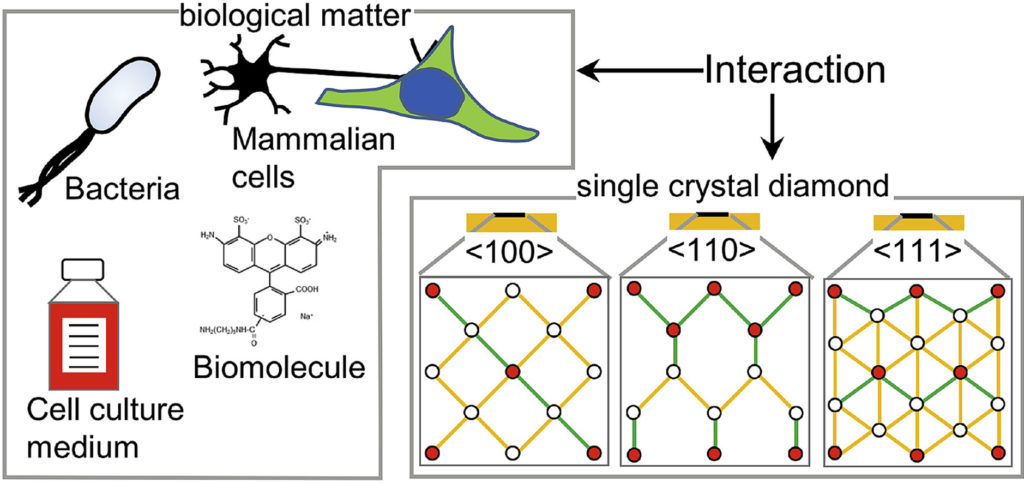 |
| Graphical Abstract: The preferential adsorption of biological matter on oriented diamond surfaces. |
Diamond has been a popular material for a variety of biological applications due to its favorable chemical, optical, mechanical and biocompatible properties. While the lattice orientation of crystalline material is known to alter the interaction between solids and biological materials, the effect of diamond’s crystal orientation on biological applications is completely unknown. Here, we experimentally evaluate the influence of the crystal orientation by investigating the interaction between the <100>, <110> and <111> surfaces of the single crystal diamond with biomolecules, cell culture medium, mammalian cells and bacteria. We show that the crystal orientation significantly alters these biological interactions. Most surprising is the two orders of magnitude difference in the number of bacteria adhering on <111> surface compared to <100> surface when both the surfaces were maintained under the same condition. We also observe differences in how small biomolecules attach to the surfaces. Neurons or HeLa cells on the other hand do not have clear preferences for either of the surfaces. To explain the observed differences, we theoretically estimated the surface charge for these three low index diamond surfaces and followed by the surface composition analysis using x-ray photoelectron spectroscopy (XPS). We conclude that the differences in negative surface charge, atomic composition and functional groups of the different surface orientations lead to significant variations in how the single crystal diamond surface interacts with the studied biological entities.
Permanent link to this article: https://dannyvanpoucke.be/paper_diamondromanaorientation2020-en/
| Authors: |
Mohammadreza Hosseini, Danny E. P. Vanpoucke, Paolo Giannozzi, Masoud Berahman and Nasser Hadipour |
| Journal: |
RSC Adv. 10, 4786-4794 (2020)
|
| doi: |
10.1039/C9RA09196C |
| IF(2019): |
3.119 |
| export: |
bibtex |
| pdf: |
<RSC Adv.> (Open Access) |
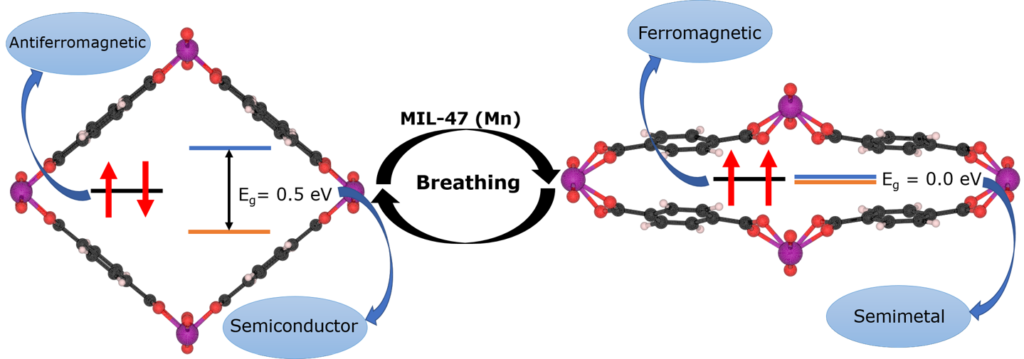 |
| Graphical Abstract: The breathing MIL-47(Mn) Metal-Organic Framework. Upon breathing, the electronic structure of this MOF undergoes a transition from an anti-ferromagnetic semiconductor, to a ferromagnetic semi-metal. |
The structural, electronic and magnetic properties of the MIL-47(Mn) metal–organic framework are investigated using first principles calculations. We find that the large-pore structure is the ground state of this material. We show that upon transition from the large-pore to the narrow-pore structure, the magnetic ground-state configuration changes from antiferromagnetic to ferromagnetic, consistent with the computed values of the intra-chain coupling constant. Furthermore, the antiferromagnetic and ferromagnetic configuration phases have intrinsically different electronic behavior: the former is semiconducting, the latter is a metal or half-metal. The change of electronic properties during breathing posits MIL-47(Mn) as a good candidate for sensing and other applications. Our calculated electronic band structure for MIL-47(Mn) presents a combination of flat dispersionless and strongly dispersive regions in the valence and conduction bands, indicative of quasi-1D electronic behavior. The spin coupling constants are obtained by mapping the total energies onto a spin Hamiltonian. The inter-chain coupling is found to be at least one order of magnitude smaller than the intra-chain coupling for both large and narrow pores. Interestingly, the intra-chain coupling changes sign and becomes five times stronger going from the large pore to the narrow pore structure. As such MIL-47(Mn) could provide unique opportunities for tunable low-dimensional magnetism in transition metal oxide systems.
Permanent link to this article: https://dannyvanpoucke.be/paper_mil47mn_moh-en/




