Tag: Hirshfeld-I
| Authors: |
Emanuele Bosoni, Louis Beal, Marnik Bercx, Peter Blaha, Stefan Blügel, Jens Bröder, Martin Callsen, Stefaan Cottenier, Augustin Degomme, Vladimir Dikan, Kristjan Eimre, Espen Flage-Larsen, Marco Fornari, Alberto Garcia, Luigi Genovese, Matteo Giantomassi, Sebastiaan P. Huber, Henning Janssen, Georg Kastlunger, Matthias Krack, Georg Kresse, Thomas D. Kühne, Kurt Lejaeghere, Georg K. H. Madsen, Martijn Marsman, Nicola Marzari, Gregor Michalicek, Hossein Mirhosseini, Tiziano M. A. Müller, Guido Petretto, Chris J. Pickard, Samuel Poncé, Gian-Marco Rignanese, Oleg Rubel, Thomas Ruh, Michael Sluydts, Danny E.P. Vanpoucke, Sudarshan Vijay, Michael Wolloch, Daniel Wortmann, Aliaksandr V. Yakutovich, Jusong Yu, Austin Zadoks, Bonan Zhu, and Giovanni Pizzi |
| Journal: |
Nature Reviews Physics 6(1), 45-58 (2024) |
| doi: |
10.1038/s42254-023-00655-3 |
| IF(2021): |
36.273 |
| export: |
bibtex |
| pdf: |
<NatRevPhys>
<ArXiv:2305.17274> |
 |
| Graphical Abstract: “We hope our dataset will be a reference for the field for years to come,” says Giovanni Pizzi, leader of the Materials Software and Data Group at the Paul Scherrer Institute PSI, who led the study. (Image: Paul Scherrer Insitute / Giovanni Pizzi) |
Density-functional theory methods and codes adopting periodic boundary conditions are extensively used in condensed matter physics and materials science research. In 2016, their precision (how well properties computed with different codes agree among each other) was systematically assessed on elemental crystals: a first crucial step to evaluate the reliability of such computations. In this Expert Recommendation, we discuss recommendations for verification studies aiming at further testing precision and transferability of density-functional-theory computational approaches and codes. We illustrate such recommendations using a greatly expanded protocol covering the whole periodic table from Z = 1 to 96 and characterizing 10 prototypical cubic compounds for each element: four unaries and six oxides, spanning a wide range of coordination numbers and oxidation states. The primary outcome is a reference dataset of 960 equations of state cross-checked between two all-electron codes, then used to verify and improve nine pseudopotential-based approaches. Finally, we discuss the extent to which the current results for total energies can be reused for different goals.
Permanent link to this article: https://dannyvanpoucke.be/paper-aiidaconsortium2023-en/
| Authors: |
Ahmed M. Rozza, Danny E. P. Vanpoucke, Eva-Maria Krammer, Julie Bouckaert, Ralf Blossey, Marc F. Lensink, Mary Jo Ondrechen, Imre Bakó, Julianna Oláh, and Goedele Roos |
| Journal: |
Journal of Molecular Liquids 384, 122172 (2023) |
| doi: |
10.1016/j.molliq.2023.122172 |
| IF(2021): |
6.633 |
| export: |
bibtex |
| pdf: |
<JMolLiq> |
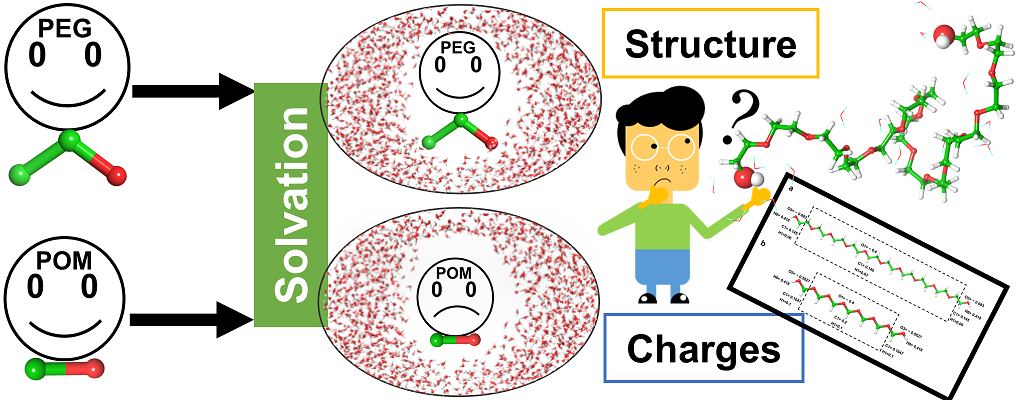 |
| Graphical Abstract: PEG or POM, similar in structure though very different in their solvation. Is this due to structure or charge(distribution)? |
Non-toxic, chemically inert, organic polymers as polyethylene glycol (PEG) and polyoxymethylene (POM) have versatile applications in basic research, industry and pharmacy. In this work, we aim to characterize the hydration structure of PEG and POM oligomers by exploring how the solute disturbs the water structure compared to the bulk solvent and how the solute chain interacts with the solvent. We explore the effect of (i) the C-C-O (PEG) versus CO (POM) constitution of the chain and (ii) chain length. To this end, MD simulations followed by clustering and topological analysis of the hydration network, as well as by quantum
mechanical calculations of atomic charges are used. We show that the hydration varies with chain conformation and length. The degree of folding of the chain impacts its degree of solvation, which is measurable by different parameters as for example the number of water molecules in the first solvation shell and the solvent accessible surface. Atomic charges calculated on the oligomers in gas phase are stable throughout conformation and chain length and seem not to determine solvation. Hydration however induces charge transfer from the solute molecule to the solvent, which depends on the degree of hydration.
Permanent link to this article: https://dannyvanpoucke.be/paper-roosg_peg-en/
| Authors: |
Viraj Damle, Kaiqi Wu, Oreste De Luca, Natalia Ortí-Casañ, Neda Norouzi, Aryan Morita, Joop de Vries, Hans Kaper, Inge Zuhorn, Ulrich Eisel, Danny E.P. Vanpoucke, Petra Rudolf, and Romana Schirhagl, |
| Journal: |
Carbon 162, 1-12 (2020) |
| doi: |
10.1016/j.carbon.2020.01.115 |
| IF(2019): |
8.821 |
| export: |
bibtex |
| pdf: |
<Carbon> (Open Access) |
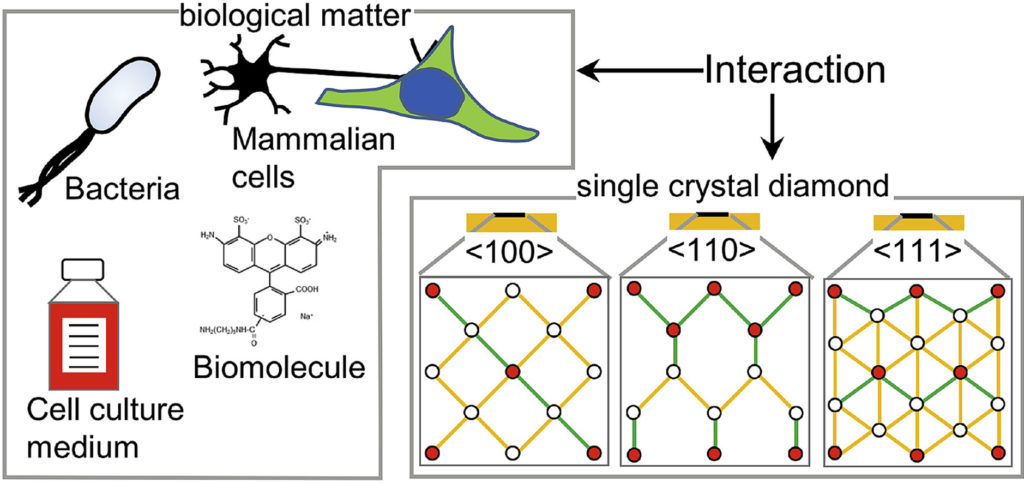 |
| Graphical Abstract: The preferential adsorption of biological matter on oriented diamond surfaces. |
Diamond has been a popular material for a variety of biological applications due to its favorable chemical, optical, mechanical and biocompatible properties. While the lattice orientation of crystalline material is known to alter the interaction between solids and biological materials, the effect of diamond’s crystal orientation on biological applications is completely unknown. Here, we experimentally evaluate the influence of the crystal orientation by investigating the interaction between the <100>, <110> and <111> surfaces of the single crystal diamond with biomolecules, cell culture medium, mammalian cells and bacteria. We show that the crystal orientation significantly alters these biological interactions. Most surprising is the two orders of magnitude difference in the number of bacteria adhering on <111> surface compared to <100> surface when both the surfaces were maintained under the same condition. We also observe differences in how small biomolecules attach to the surfaces. Neurons or HeLa cells on the other hand do not have clear preferences for either of the surfaces. To explain the observed differences, we theoretically estimated the surface charge for these three low index diamond surfaces and followed by the surface composition analysis using x-ray photoelectron spectroscopy (XPS). We conclude that the differences in negative surface charge, atomic composition and functional groups of the different surface orientations lead to significant variations in how the single crystal diamond surface interacts with the studied biological entities.
Permanent link to this article: https://dannyvanpoucke.be/paper_diamondromanaorientation2020-en/
| Authors: |
Jarod J. Wolffis, Danny E. P. Vanpoucke, Amit Sharma, Keith V. Lawler, and Paul M. Forster |
| Journal: |
Microporous Mesoporous Mater. 277, 184-196 (2019) |
| doi: |
10.1016/j.micromeso.2018.10.028 |
| IF(2019): |
4.551 |
| export: |
bibtex |
| pdf: |
<MicroporousMesoporousMater> |
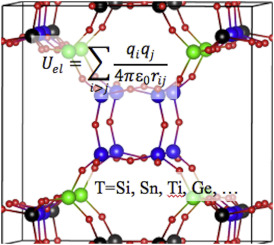 |
| Graphical Abstract: Partial charges in zeolites for force fields. |
Partial atomic charge, which determines the magnitude of the Coulombic non-bonding interaction, represents a critical parameter in molecular mechanics simulations. Partial charges may also be used as a measure of physical properties of the system, i.e. covalency, acidic/catalytic sites, etc. A range of methods, both empirical and ab initio, exist for calculating partial charges in a given solid, and several of them are compared here for siliceous (pure silica) zeolites. The relationships between structure and the predicted partial charge are examined. The predicted partial charges from different methods are also compared with related experimental observations, showing that a few of the methods offer some guidance towards identifying the T-sites most likely to undergo substitution or for proton localization in acidic framework forms. Finally, we show that assigning unique calculated charges to crystallographically unique framework atoms makes an appreciable difference in simulating predicting N2 and O2 adsorption with common dispersion-repulsion parameterizations.
Permanent link to this article: https://dannyvanpoucke.be/paper_hizeolites_2018-en/
| Authors: |
Danny E. P. Vanpoucke |
| Journal: |
J. Phys. Chem. C 121(14), 8014-8022 (2017) |
| doi: |
10.1021/acs.jpcc.7b01491 |
| IF(2017): |
4.484 |
| export: |
bibtex |
| pdf: |
<J.Phys.Chem.C> |
 upon OH-functionalization of the BDC linker.") |
| Graphical Abstract: Evolution of the electronic band structure of MIL-47(V) upon OH-functionalization of the BDC linker. The π-orbital of the BDC linker splits upon functionalisation, and the split-off π-band moves up into the band gap, effectively reducing the latter. |
Metal–organic frameworks (MOFs) have gained much interest due to their intrinsic tunable nature. In this work, we study how linker functionalization modifies the electronic structure of the host MOF, more specifically, the MIL-47(V)-R (R = −F, −Cl, −Br, −OH, −CH3, −CF3, and −OCH3). It is shown that the presence of a functional group leads to a splitting of the π orbital on the linker. Moreover, the upward shift of the split-off π-band correlates well with the electron-withdrawing/donating nature of the functional groups. For halide functional groups the presence of lone-pair back-donation is corroborated by calculated Hirshfeld-I charges. In the case of the ferromagnetic configuration of the host MIL-47(V+IV) material a half-metal to insulator transition is noted for the −Br, −OCH3, and −OH functional groups, while for the antiferromagnetic configuration only the hydroxy group results in an effective reduction of the band gap.
Permanent link to this article: https://dannyvanpoucke.be/mof-mil47-linkerfunct-en/
| Authors: |
Arthur De Vos, Kurt Lejaeghere, Danny E. P. Vanpoucke, Jonas J. Joos, Philippe F. Smet, and Karen Hemelsoet |
| Journal: |
Inorg. Chem. 55(5), 2402-2412 (2016) |
| doi: |
10.1021/acs.inorgchem.5b02805 |
| IF(2016): |
4.857 |
| export: |
bibtex |
| pdf: |
<Inorg.Chem> |
 Ball-and-stick model of zinc gallate (right) density of states of Cr doped zinc gallate.") |
| Graphical Abstract: First-principles simulations on zinc gallate solid phosphors (ZGO) containing a chromium dopant and antisite defects (left) rationalize the attractive interactions between the various elements. A large number of antisite pair configurations is investigated and compared with isolated antisite defects. Defect energies point out the stability of the antisite defects in ZGO. Local structural distortions are reported, and charge transfer mechanisms are analyzed based on theoretical density of states (right) and Hirshfeld-I charges. |
Zinc gallate doped with chromium is a recently developed near-infrared emitting persistent phosphor, which is now extensively studied for in vivo bioimaging and security applications. The precise mechanism of this persistent luminescence relies on defects, in particular, on antisite defects and antisite pairs. A theoretical model combining the solid host, the dopant, and/or antisite defects is constructed to elucidate the mutual interactions in these complex materials. Energies of formation as well as dopant, and defect energies are calculated through density-functional theory simulations of large periodic supercells. The calculations support the chromium substitution on the slightly distorted octahedrally coordinated gallium site, and additional energy levels are introduced in the band gap of the host. Antisite pairs are found to be energetically favored over isolated antisites due to significant charge compensation as shown by calculated Hirshfeld-I charges. Significant structural distortions are found around all antisite defects. The local Cr surrounding is mainly distorted due to a ZnGa antisite. The stability analysis reveals that the distance between both antisites dominates the overall stability picture of the material containing the Cr dopant and an antisite pair. The findings are further rationalized using calculated densities of states and Hirshfeld-I charges.
Permanent link to this article: https://dannyvanpoucke.be/paper2016_inorgchemzgodoping-en/
| Authors: |
Danny E. P. Vanpoucke, Julianna Oláh, Frank De Proft, Veronique Van Speybroeck, and Goedele Roos |
| Journal: |
J. Chem. Inf. Model. 55(3), 564-571 (2015) |
| doi: |
10.1021/ci5006417 |
| IF(2015): |
3.657 |
| export: |
bibtex |
| pdf: |
<J.Chem.Inf.Model.> |
 |
| Graphical Abstract: Graphical Abstract: The influence of the cluster size and water presence on the atomic charge of active and inactive sites in Bio-molecules. |
Atomic charges are a key concept to give more insight into the electronic structure and chemical reactivity. The Hirshfeld-I partitioning scheme applied to the model protein human 2-cysteine peroxiredoxin thioredoxin peroxidase B is used to investigate how large a protein fragment needs to be in order to achieve convergence of the atomic charge of both, neutral and negatively charged residues. Convergence in atomic charges is rapidly reached for neutral residues, but not for negatively charged ones. This study pinpoints difficulties on the road towards accurate modeling of negatively charged residues of large bio-molecular systems in a multiscale approach.
Permanent link to this article: https://dannyvanpoucke.be/paper2015_jcheminfo-en/
| Authors: |
Danny E. P. Vanpoucke, Jan W. Jaeken, Stijn De Baerdemacker, Kurt Lejaeghere
and Veronique Van Speybroeck |
| Journal: |
Beilstein J. Nanotechnol. 5, 1738-1748 (2014) |
| doi: |
10.3762/bjnano.5.184 |
| IF(2014): |
2.670 |
| export: |
bibtex |
| pdf: |
<Beilstein> (open access) |
 Spin density of anti-ferromagnetic MIL-47(V) with ferromagnetic chains. (right) Electronic band structure and density of states.") |
| Graphical Abstract: The MIL-47(V) MOF has one unpaired electron per V site. As a result, different spin configurations are possible, several of which lead to an anti-ferromagnetic state. The spin density of an antiferromagnetic state, containing only ferromagnetic chains is shown on the left. On the right, the electronic band structure of the same system is presented. |
The geometric and electronic structure of the MIL-47(V) metal-organic framework (MOF) is investigated by using ab initio density functional theory (DFT) calculations. Special focus is placed on the relation between the spin configuration and the properties of the MOF. The ground state is found to be antiferromagnetic, with an equilibrium volume of 1554.70 Å3. The transition pressure of the pressure-induced large-pore-to-narrow-pore phase transition is calculated to be 82 MPa and 124 MPa for systems with ferromagnetic and antiferromagnetic chains, respectively. For a mixed system, the transition pressure is found to be a weighted average of the ferromagnetic and antiferromagnetic transition pressures. Mapping DFT energies onto a simple-spin Hamiltonian shows both the intra- and inter-chain coupling to be antiferromagnetic, with the latter coupling constant being two orders of magnitude smaller than the former, suggesting the MIL-47(V) to present quasi-1D behavior. The electronic structure of the different spin configurations is investigated and it shows that the band gap position varies strongly with the spin configuration. The valence and conduction bands show a clear V d-character. In addition, these bands are flat in directions orthogonal to VO6 chains, while showing dispersion along the the direction of the VO6 chains, similar as for other quasi-1D materials.
Permanent link to this article: https://dannyvanpoucke.be/paper2014_mil47beil-en/
| Authors: |
Danny E. P. Vanpoucke, Stefaan Cottenier, Veronique Van Speybroeck, Isabel Van Driessche, and Patrick Bultinck |
| Journal: |
J. Am. Ceram. Soc. 97(1), 258-266 (2014) |
| doi: |
10.1111/jace.12650 |
| IF(2014): |
2.610 |
| export: |
bibtex |
| pdf: |
<J.Am.Ceram.Soc.> <arXiv> |
Fluorite CeO2 doped with group IV elements is studied within the density functional theory (DFT) and DFT + U framework. Concentration-dependent formation energies are calculated for Ce1−xZxO2 (Z = C, Si, Ge, Sn, Pb, Ti, Zr, Hf) with 0 ≤ x ≤ 0.25 and a roughly decreasing trend with ionic radius is observed. The influence of the valence and near valence electronic configuration is discussed, indicating the importance of filled d and f shells near the Fermi level for all properties investigated. A clearly different behavior of group IVa and IVb dopants is observed: the former are more suitable for surface modifications and the latter are more suitable for bulk modifications. For the entire set of group IV dopants, there exists an inverse relation between the change, due to doping, of the bulk modulus, and the thermal expansion coefficients. Hirshfeld-I atomic charges show that charge-transfer effects due to doping are limited to the nearest-neighbor oxygen atoms.
Permanent link to this article: https://dannyvanpoucke.be/paper2014_ceo2group4-en/
| Authors: |
Danny E. P. Vanpoucke |
| Journal: |
J. Comput. Chem. 34(5), i-ii (2013) |
| doi: |
10.1002/jcc.23239 |
| IF(2013): |
3.601 |
| export: |
bibtex |
| pdf: |
<J.Comput.Chem.> |
The image shows an isosurface of Hirshfeld-I “atoms in molecules” for Ti-doped CeO2, taken at an electron density of 0.03e/Å3, as presented by Danny E. P. Vanpoucke, Patrick Bultinck, and Isabel Van Driessche on page 405. The cubic Ce0.75Ti0.25O2 unit cell is shown along the 111 direction. The different atoms are still clearly distinguishable at this iso-surface level, and show the Ti atom in the corners to be much smaller than the Ce atoms on the sides. In this issue, this implementation of the Hirshfeld- I method for solids is published back to back with a Comment from Thomas A. Manz and the authors’ Reply.

Permanent link to this article: https://dannyvanpoucke.be/paper2013_hicover-en/




