Tag: electronic band structure
| Authors: |
Danny E. P. Vanpoucke |
| Journal: |
J. Phys. Chem. C 121(14), 8014-8022 (2017) |
| doi: |
10.1021/acs.jpcc.7b01491 |
| IF(2017): |
4.484 |
| export: |
bibtex |
| pdf: |
<J.Phys.Chem.C> |
 upon OH-functionalization of the BDC linker.") |
| Graphical Abstract: Evolution of the electronic band structure of MIL-47(V) upon OH-functionalization of the BDC linker. The π-orbital of the BDC linker splits upon functionalisation, and the split-off π-band moves up into the band gap, effectively reducing the latter. |
Metal–organic frameworks (MOFs) have gained much interest due to their intrinsic tunable nature. In this work, we study how linker functionalization modifies the electronic structure of the host MOF, more specifically, the MIL-47(V)-R (R = −F, −Cl, −Br, −OH, −CH3, −CF3, and −OCH3). It is shown that the presence of a functional group leads to a splitting of the π orbital on the linker. Moreover, the upward shift of the split-off π-band correlates well with the electron-withdrawing/donating nature of the functional groups. For halide functional groups the presence of lone-pair back-donation is corroborated by calculated Hirshfeld-I charges. In the case of the ferromagnetic configuration of the host MIL-47(V+IV) material a half-metal to insulator transition is noted for the −Br, −OCH3, and −OH functional groups, while for the antiferromagnetic configuration only the hydroxy group results in an effective reduction of the band gap.
Permanent link to this article: https://dannyvanpoucke.be/mof-mil47-linkerfunct-en/
| Authors: |
Arthur De Vos, Kurt Lejaeghere, Danny E. P. Vanpoucke, Jonas J. Joos, Philippe F. Smet, and Karen Hemelsoet |
| Journal: |
Inorg. Chem. 55(5), 2402-2412 (2016) |
| doi: |
10.1021/acs.inorgchem.5b02805 |
| IF(2016): |
4.857 |
| export: |
bibtex |
| pdf: |
<Inorg.Chem> |
 Ball-and-stick model of zinc gallate (right) density of states of Cr doped zinc gallate.") |
| Graphical Abstract: First-principles simulations on zinc gallate solid phosphors (ZGO) containing a chromium dopant and antisite defects (left) rationalize the attractive interactions between the various elements. A large number of antisite pair configurations is investigated and compared with isolated antisite defects. Defect energies point out the stability of the antisite defects in ZGO. Local structural distortions are reported, and charge transfer mechanisms are analyzed based on theoretical density of states (right) and Hirshfeld-I charges. |
Zinc gallate doped with chromium is a recently developed near-infrared emitting persistent phosphor, which is now extensively studied for in vivo bioimaging and security applications. The precise mechanism of this persistent luminescence relies on defects, in particular, on antisite defects and antisite pairs. A theoretical model combining the solid host, the dopant, and/or antisite defects is constructed to elucidate the mutual interactions in these complex materials. Energies of formation as well as dopant, and defect energies are calculated through density-functional theory simulations of large periodic supercells. The calculations support the chromium substitution on the slightly distorted octahedrally coordinated gallium site, and additional energy levels are introduced in the band gap of the host. Antisite pairs are found to be energetically favored over isolated antisites due to significant charge compensation as shown by calculated Hirshfeld-I charges. Significant structural distortions are found around all antisite defects. The local Cr surrounding is mainly distorted due to a ZnGa antisite. The stability analysis reveals that the distance between both antisites dominates the overall stability picture of the material containing the Cr dopant and an antisite pair. The findings are further rationalized using calculated densities of states and Hirshfeld-I charges.
Permanent link to this article: https://dannyvanpoucke.be/paper2016_inorgchemzgodoping-en/
| Authors: |
Bart Bueken, Frederik Vermoortele, Danny E. P. Vanpoucke, Helge Reinsch, Chih-Chin Tsou, Pieterjan Valvekens, Trees De Baerdemaeker, Rob Ameloot, Christine E. A. Kirschhock, Veronique Van Speybroeck, James M. Mayer and Dirk De Vos |
| Journal: |
Angew. Chem. Int. Ed. 54(47), 13912-13917 (2015) |
| doi: |
10.1002/anie.201505512 |
| IF(2015): |
11.705 |
| export: |
bibtex |
| pdf: |
<Angew.Chem.Int.Ed.> |
The synthesis of titanium-carboxylate metal-organic frameworks (MOFs) is hampered by the high reactivity of the commonly employed alkoxide precursors. Here, we present an innovative approach to Ti-based MOFs using titanocene dichloride to synthesize COK-69, the first breathing Ti-MOF built up of trans-1,4- cyclohexanedicarboxylate linkers and an unprecedented [TiIV3(µ3-O)(O)2(COO)6] cluster. The photoactive properties of COK-69 were investigated in-depth by proton-coupled electron transfer experiments, which revealed that up to one TiIV per cluster can be photoreduced to TiIII, while preserving the structural integrity of the framework. From molecular modeling, the electronic structure of COK-69 was determined and a band gap of 3.77 eV was found.
Permanent link to this article: https://dannyvanpoucke.be/paper2015_aniecok69-en/
| Authors: |
Kevin Hendrickx, Danny E.P. Vanpoucke, Karen Leus, Kurt Lejaeghere,
Andy Van Yperen-De Deyne, Veronique Van Speybroeck, Pascal Van Der
Voort, and Karen Hemelsoet |
| Journal: |
Inorg. Chem. 54(22), 10701-10710 (2015) |
| doi: |
10.1021/acs.inorgchem.5b01593 |
| IF(2015): |
4.820 |
| export: |
bibtex |
| pdf: |
<Inorg.Chem.> |
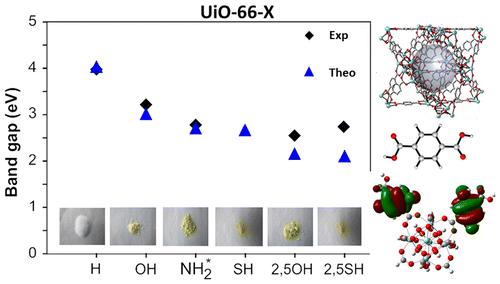
Linker-functionalization of UiO-66 modifies the optical band gap and thus the color of the MOF.
A combined theoretical and experimental study is performed in order to elucidate the eff�ects of linker functional groups on the photoabsorption properties of UiO-66-type materials. This study, in which both mono- and di-functionalized linkers (with X= -OH, -NH2, -SH) are studied, aims to obtain a more complete picture on the choice of functionalization. Static Time-Dependent Density Functional Theory (TD-DFT) calculations combined with Molecular Dynamics simulations are performed on the linkers and compared to experimental UV/VIS spectra, in order to understand the electronic eff�ects governing the absorption spectra. Di-substituted linkers show larger shifts compared to mono-substituted variants, making them promising candidates for further study as photocatalysts. Next, the interaction between the linker and the inorganic part of the framework is theoretically investigated using a cluster model. The proposed Ligand-to-Metal-Charge Transfer (LMCT) is theoretically observed and is influenced by the differences in functionalization. Finally, computed electronic properties of the periodic UiO-66 materials reveal that the band gap can be altered by linker functionalization and ranges from 4.0 down to 2.2 eV. Study of the periodic Density of States (DOS) allows to explain the band gap modulations of the framework in terms of a functionalization-induced band in the band gap of the original UiO-66 host.
Permanent link to this article: https://dannyvanpoucke.be/paper2015_inorgchemuio66-en/

