Category: 2015
| Authors: |
Bart Bueken, Frederik Vermoortele, Danny E. P. Vanpoucke, Helge Reinsch, Chih-Chin Tsou, Pieterjan Valvekens, Trees De Baerdemaeker, Rob Ameloot, Christine E. A. Kirschhock, Veronique Van Speybroeck, James M. Mayer and Dirk De Vos |
| Journal: |
Angew. Chem. Int. Ed. 54(47), 13912-13917 (2015) |
| doi: |
10.1002/anie.201505512 |
| IF(2015): |
11.705 |
| export: |
bibtex |
| pdf: |
<Angew.Chem.Int.Ed.> |
The synthesis of titanium-carboxylate metal-organic frameworks (MOFs) is hampered by the high reactivity of the commonly employed alkoxide precursors. Here, we present an innovative approach to Ti-based MOFs using titanocene dichloride to synthesize COK-69, the first breathing Ti-MOF built up of trans-1,4- cyclohexanedicarboxylate linkers and an unprecedented [TiIV3(µ3-O)(O)2(COO)6] cluster. The photoactive properties of COK-69 were investigated in-depth by proton-coupled electron transfer experiments, which revealed that up to one TiIV per cluster can be photoreduced to TiIII, while preserving the structural integrity of the framework. From molecular modeling, the electronic structure of COK-69 was determined and a band gap of 3.77 eV was found.
Permanent link to this article: https://dannyvanpoucke.be/paper2015_aniecok69-en/
| Authors: |
Kevin Hendrickx, Danny E.P. Vanpoucke, Karen Leus, Kurt Lejaeghere,
Andy Van Yperen-De Deyne, Veronique Van Speybroeck, Pascal Van Der
Voort, and Karen Hemelsoet |
| Journal: |
Inorg. Chem. 54(22), 10701-10710 (2015) |
| doi: |
10.1021/acs.inorgchem.5b01593 |
| IF(2015): |
4.820 |
| export: |
bibtex |
| pdf: |
<Inorg.Chem.> |
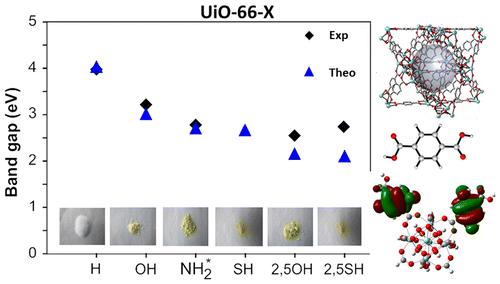
Linker-functionalization of UiO-66 modifies the optical band gap and thus the color of the MOF.
A combined theoretical and experimental study is performed in order to elucidate the eff�ects of linker functional groups on the photoabsorption properties of UiO-66-type materials. This study, in which both mono- and di-functionalized linkers (with X= -OH, -NH2, -SH) are studied, aims to obtain a more complete picture on the choice of functionalization. Static Time-Dependent Density Functional Theory (TD-DFT) calculations combined with Molecular Dynamics simulations are performed on the linkers and compared to experimental UV/VIS spectra, in order to understand the electronic eff�ects governing the absorption spectra. Di-substituted linkers show larger shifts compared to mono-substituted variants, making them promising candidates for further study as photocatalysts. Next, the interaction between the linker and the inorganic part of the framework is theoretically investigated using a cluster model. The proposed Ligand-to-Metal-Charge Transfer (LMCT) is theoretically observed and is influenced by the differences in functionalization. Finally, computed electronic properties of the periodic UiO-66 materials reveal that the band gap can be altered by linker functionalization and ranges from 4.0 down to 2.2 eV. Study of the periodic Density of States (DOS) allows to explain the band gap modulations of the framework in terms of a functionalization-induced band in the band gap of the original UiO-66 host.
Permanent link to this article: https://dannyvanpoucke.be/paper2015_inorgchemuio66-en/
| Authors: |
Danny E. P. Vanpoucke, Kurt Lejaeghere, Veronique Van Speybroeck, Michel Waroquier, and An
Ghysels |
| Journal: |
J. Phys. Chem. C 119(41), 23752-23766 (2015) |
| doi: |
10.1021/acs.jpcc.5b06809 |
| IF(2015): |
4.509 |
| export: |
bibtex |
| pdf: |
<J.Phys.Chem.C> |
 Metal-Organic Framework.") |
| Graphical Abstract: Pulay stresses complicate the structure optimization of the breathing MIL-47(V) Metal-Organic Framework. |
Modeling the flexibility of metal–organic frameworks (MOFs) requires the computation of mechanical properties from first principles, e.g., for screening of materials in a database, for gaining insight into structural transformations, and for force field development. However, this paper shows that computations with periodic density functional theory are challenged by the flexibility of these materials: guidelines from experience with standard solid-state calculations cannot be simply transferred to flexible porous frameworks. Our test case, the MIL-47(V) material, has a large-pore and a narrow-pore shape. The effect of Pulay stress (cf. Pulay forces) leads to drastic errors for a simple structure optimization of the flexible MIL-47(V) material. Pulay stress is an artificial stress that tends to lower the volume and is caused by the finite size of the plane wave basis set. We have investigated the importance of this Pulay stress, of symmetry breaking, and of k-point sampling on (a) the structure optimization and (b) mechanical properties such as elastic constants and bulk modulus, of both the large-pore and narrow-pore structure of MIL-47(V). We found that, in the structure optimization, Pulay effects should be avoided by using a fitting procedure, in which an equation of state E(V) (EOS) is fit to a series of energy versus volume points. Manual symmetry breaking could successfully lower the energy of MIL-47(V) by distorting the vanadium–oxide distances in the vanadyl chains and by rotating the benzene linkers. For the mechanical properties, the curvature of the EOS curve was compared with the Reuss bulk modulus, derived from the elastic tensor in the harmonic approximation. Errors induced by anharmonicity, the eggbox effect, and Pulay effects propagate into the Reuss modulus. The strong coupling of the unit cell axes when the unit cell deforms expresses itself in numerical instability of the Reuss modulus. For a flexible material, it is therefore advisible to resort to the EOS fit procedure.
Permanent link to this article: https://dannyvanpoucke.be/paper2015_accuratemofs-en/
| Authors: |
Thomas Bogaerts, Louis Vanduyfhuys, Danny E. P. Vanpoucke, Jelle Wieme,
Michel Waroquier, Pascal van der Voort and Veronique van Speybroeck |
| Journal: |
Cryst. Eng. Comm. 17(45), 8612-8622 (2015) |
| doi: |
10.1039/c5ce01388g |
| IF(2015): |
3.849 |
| export: |
bibtex |
| pdf: |
<Cryst.Eng.Comm.> |
 |
| Graphical Abstract: Which model represents the experimental XRD-spectra best? Ferromagnetic or anti-ferromagnetic chains? With of without offset? |
The structural characterization of complex crystalline materials such as metal organic frameworks can prove a very difficult challenge both for experimentalists as for theoreticians. From theory, the flat potential energy surface of these highly flexible structures often leads to different geometries that are energetically very close to each other. In this work a distinction between various computationally determined structures is made by comparing experimental and theoretically derived X-ray diffractograms which are produced from the materials geometry. The presented approach allows to choose the most appropriate geometry of a MIL-47(V) MOF and even distinguish between different electronic configurations that induce small structural changes. Moreover the techniques presented here are used to verify the applicability of a newly developed force field for this material. The discussed methodology is of significant importance for modelling studies where accurate geometries are crucial, such as mechanical properties and adsorption of guest molecules.
Permanent link to this article: https://dannyvanpoucke.be/paper2015_xrd_crystendcomm-en/
| Authors: |
Danny E. P. Vanpoucke, Julianna Oláh, Frank De Proft, Veronique Van Speybroeck, and Goedele Roos |
| Journal: |
J. Chem. Inf. Model. 55(3), 564-571 (2015) |
| doi: |
10.1021/ci5006417 |
| IF(2015): |
3.657 |
| export: |
bibtex |
| pdf: |
<J.Chem.Inf.Model.> |
 |
| Graphical Abstract: Graphical Abstract: The influence of the cluster size and water presence on the atomic charge of active and inactive sites in Bio-molecules. |
Atomic charges are a key concept to give more insight into the electronic structure and chemical reactivity. The Hirshfeld-I partitioning scheme applied to the model protein human 2-cysteine peroxiredoxin thioredoxin peroxidase B is used to investigate how large a protein fragment needs to be in order to achieve convergence of the atomic charge of both, neutral and negatively charged residues. Convergence in atomic charges is rapidly reached for neutral residues, but not for negatively charged ones. This study pinpoints difficulties on the road towards accurate modeling of negatively charged residues of large bio-molecular systems in a multiscale approach.
Permanent link to this article: https://dannyvanpoucke.be/paper2015_jcheminfo-en/

