“Once upon a time, there was a young researcher studying the formation of Pt nanowires on Ge substrates using quantum mechanical simulations. The results of the experimental counterparts were excellent; they provided Scanning Tunneling Microscopy images of ridiculously high quality …but not really atomistic structural information or detailed electronic band structures. On the other hand, the calculation-software of the young researcher provided only ground state energies and electronic band structures…but no Scanning Tunneling Microscopy images. So the young researcher set out to resolve this discrepancy.”
About 15 years ago, when starting out as a fresh Ph.D. student, I faced this mismatch between what my calculations could do and what my experimental counterparts had on offer. High quality ground state energies are nice, but rather useless in an experimental context governed by meta-stable states and high temperature transitions (especially since DFT represents only 0K results). I had to find a way to connect my calculations directly to the available experimental data, which boiled down to simulating Scanning Tunneling Microscopy (STM) images.
Original Delphi program: Graphene
At that time, my programming skills were still nascent, but I felt king of the world knowing both pascal/Delphi and C/C++. I had written toy-programs in both languages, going from a text based battleships in turbo-pascal over a brick-buster game in C/C++using the djgpp compiler and allegro library to create the GUI, and many GUI programs in Delphi (e.g., the programs needed to numerically calculated Bose-Eistein condensation behavior for molecular condensates during my masters thesis). Based on those experiences, I knew that writing a GUI program was much more straight forward in Delphi. So I set out writing my STM program using Delphi in the year(s) 2005-2006**. On the right you can see an screenshot of this program, generated today 15 years later, on the electron density of graphene. The program written in windows XP, ran smoothly and without modification or required recompile on both windows 7 and the current windows 10. Not to bad, if I say so myself. Try that with a python “program” 😈 .[1]

Simulated STM image of the Pt-induced nanowires on the Ge(001) surface. Green discs indicate the atomic positions of the bulk-Ge atoms; red: Pt atoms embedded in the top surface layers; yellow: Ge atoms forming the nanowire observed by STM.
The program was designed to work for my specific use-case at the time: a germanium 001 surface, with a nice rectangular surface unit cell (see figure on the left). This has the unfortunate consequence that systems with a non-rectangular unit cell appear skewed, as is seen for the graphene example above. However, as I never needed such systems myself, no fix was ever included.
After presenting STM results in my first published paper in 2008,[2] I got some questions if it was possible to share the program. I shared the program on an as-is basis: free to use, and I hope it works for you as well, but no support.
Reading the above you may wonder: “Why didn’t you put the source on GitHub, such that other people could collaborate with you on it, and extend it and fix bugs?” The answer is rather simple (and sobering at the same time): GitHub didn’t exist yet when I wrote the program, as it was founded only in February 2008. It grew rapidly since then (surpassing SourceForge in mid 2011), but as I was working on other projects there was no time to support such a setup.
The number of people asking for the program grew steadily, and there was the nagging feeling at the back of my head that I should really clean up the code and make it cross-platform. In 2011, I had a short period when I decided to start from scratch and write the program anew in Java. Unfortunately, my available time ran out, and initial tests showed the program had a hard time reading the large charge-density files fast. So the original Delphi version remained in use being distributed to new users. By September 2012, this program developed for my own purposes had been requested by 100 researchers (which is a lot considering the boundary conditions: (1) needing atomic scale STM simulations and (2) using VASP for DFT calculations), and over 200 researchers had requested it by 2015. Currently, in January 2021, the counter indicates over 400 requests. Still the same piece of software, being used by people I never imagined would be interested on OS’s it was never designed for. Despite its simplicity, this unexpected interest makes me extremely proud. 😎
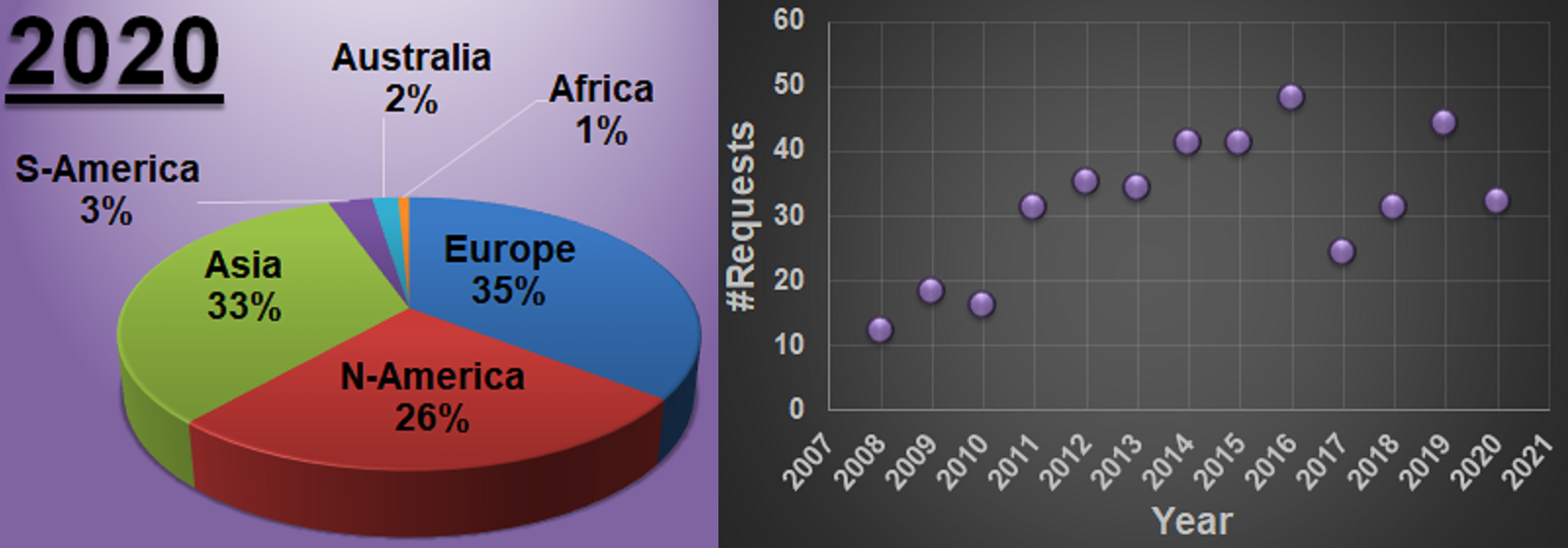
Distribution of users over the continents and evolution of requests over the years.
Thorny roses: Some issues popping up
Given the original intent of the program and its eventual use, one should not be amazed that some issues popped up over the years. However, no serious bugs were encountered (which still amazes me).
- Non-orthogonal surface units: This is the oldest known limitation of the program. It assumes a rectangular surface unit as it uses the direct grid used in the VASP CHGCAR file as a pixel grid. This suited my own purposes well, but is unfortunate for the user studying hexagonal surfaces.
- “Smart” Antivirus software (1): In the early days, I just sent a zipfile with the program and manual to new users. Unfortunately, AVs do not like people mailing executables, leading to mails being blocked. For some time the problem could be circumvented by zipping the zipfile and later even renaming the extension of the second zip round to prevent the AV of trying to look inside. I know, one should not do this and applaud the AVs for protecting their users, as people did spread trojan horses and other viruses like this back in the days. (Who clicks on those strange attachments anyhow?) So we ended up storing the program and zip online with password protection. We are not yet safe of AVs as some still complain about the risks of downloading things of the internet…but at least we are not (yet) back at the automatic shredding of the program.
- “Smart” Antivirus software (2): Did I say the program was written in Delphi? Apparently so were a lot of computer viruses and worms. (Must be a sign of being a nice and easy to use language 🙂 ) With smart AVs training on pieces of code from such fraudulent software it becomes rather hard to write any code using Delphi which has not been part of a virus…and thus your program gets flagged. Some AVs are nice enough to tell the user, and even provide an option to keep the program. Others just shred it without even mentioning it (not cool). This is unfortunately becoming more of a problem. Online multi-virus-scanners give a rather bleak picture, as can be seen below.
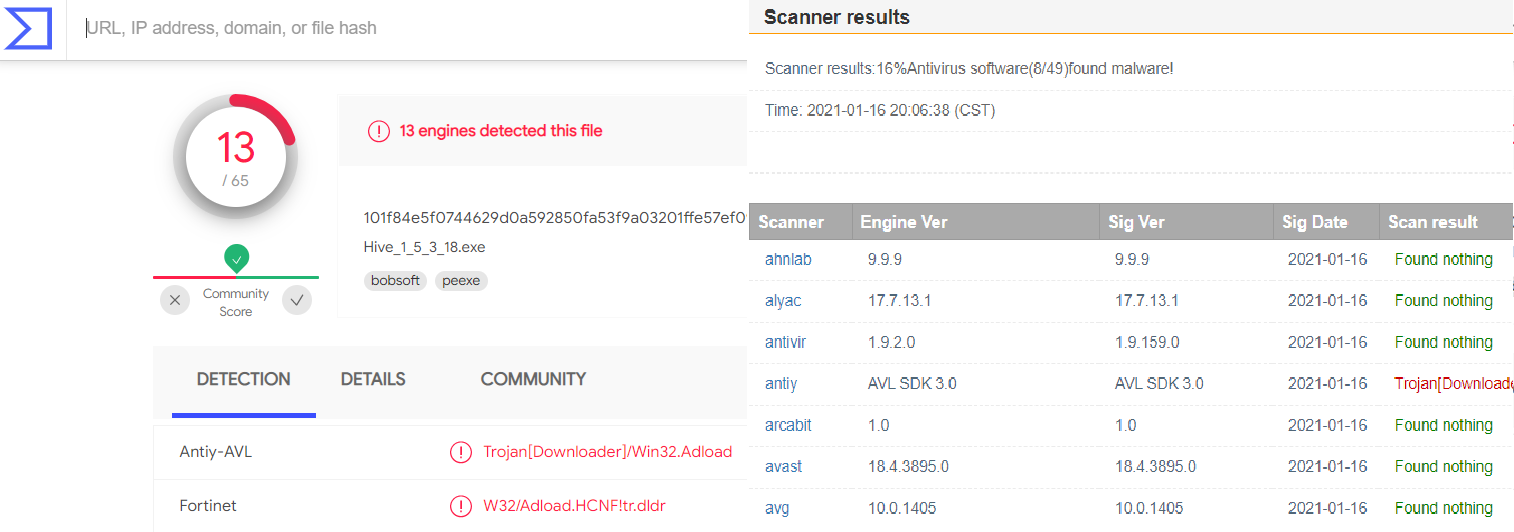
smart AVs giving false positives on the old HIVE executable.
- Windows 10: Extending on the previous, windows 10’s anti-virus protection follows suit throwing up warnings and messages of possible security threats.
- Mac and 64bit: Although the program was written for windows, it also runs smoothly in unix environments when using an emulator such as Wine, making the program available to Linux and Mac users as well. Unfortunately since the Mac OS version Catalina, Mac has dropped support for 32bit executables, making it no longer possible to run the 15 year old executable. [1] Remember that in 2006 64bit programs were new and not generally supported. Furthermore, 32bit executables tend to work smoothly on a 64bit system, they just “waste” half the memory.
The Future of HIVE-STM
Over the years, I’ve often considered it time to clean up the code, and upgrading it. Unfortunately time was always a major issue. In addition, I no longer had a working Delphi compiler so I was lured to the idea of rewriting it in a different programming language (I seriously considered reworking it in fortran, though the easy access to a GUI stopped me from doing this).
The latest issue with Macs and the zealous persecution of Delphi programs by AVs finally got me to the point of starting a full rework of the HIVE-STM program as a hobby project. The maturity of the Lazarus IDE and free-pascal compiler is an important second component. During the summer holidays of 2020, I started porting the original Delphi code to the Lazarus IDE and free-pascal. This successful port gave me the courage to continue working on it, and I am currently performing a full rewrite of the internals (so far things have gone smoothly). The new version will become available via GitHub once I am confident it is working well and a have setup a good method of keeping track of new users.
New years resolution 2021:
“Finally build a new ‘updated’ version of HIVE-STM “
References
[1] “Challenge to scientists: does your ten-year-old code still run?“, J.M. Perkel, nature technology feature, august 24th 2020.
[2] “Formation of Pt-induced Ge atomic nanowires on Pt/Ge(001)“, D.E.P. Vanpoucke & G. Brocks, Phys. Rev. B 77, 241308(R) 2008.



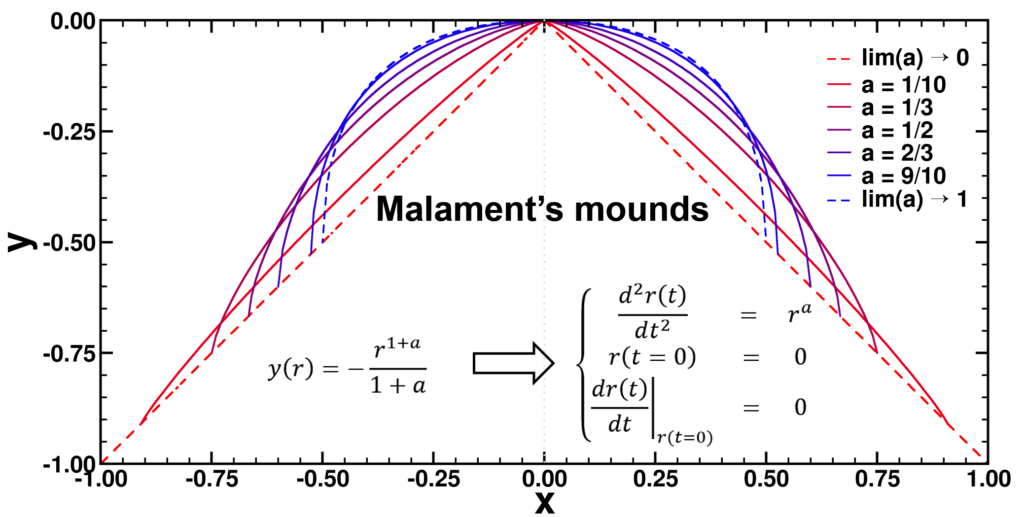
 ,
,  = 0") , and
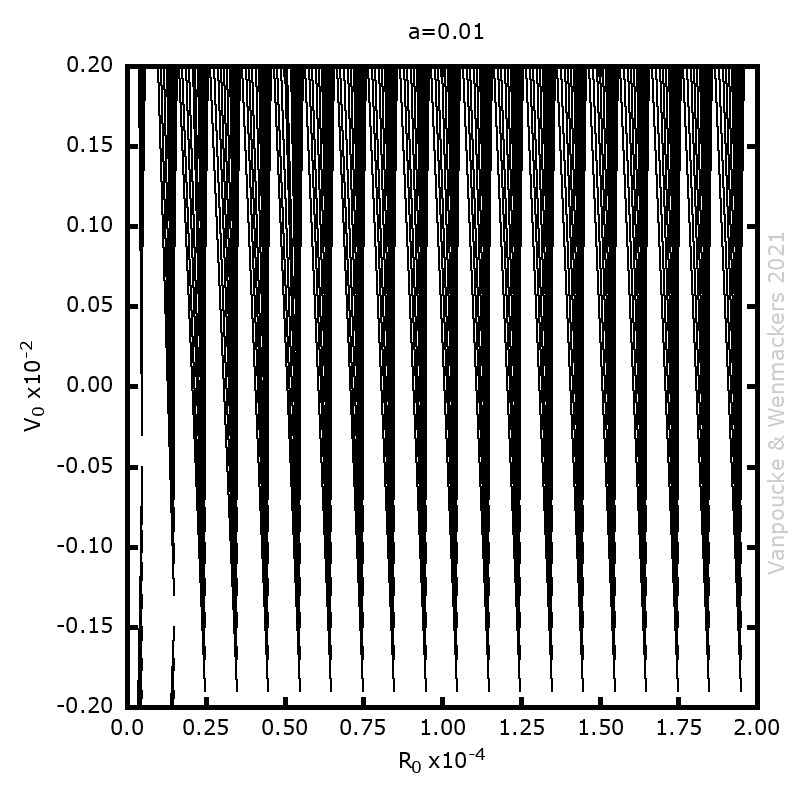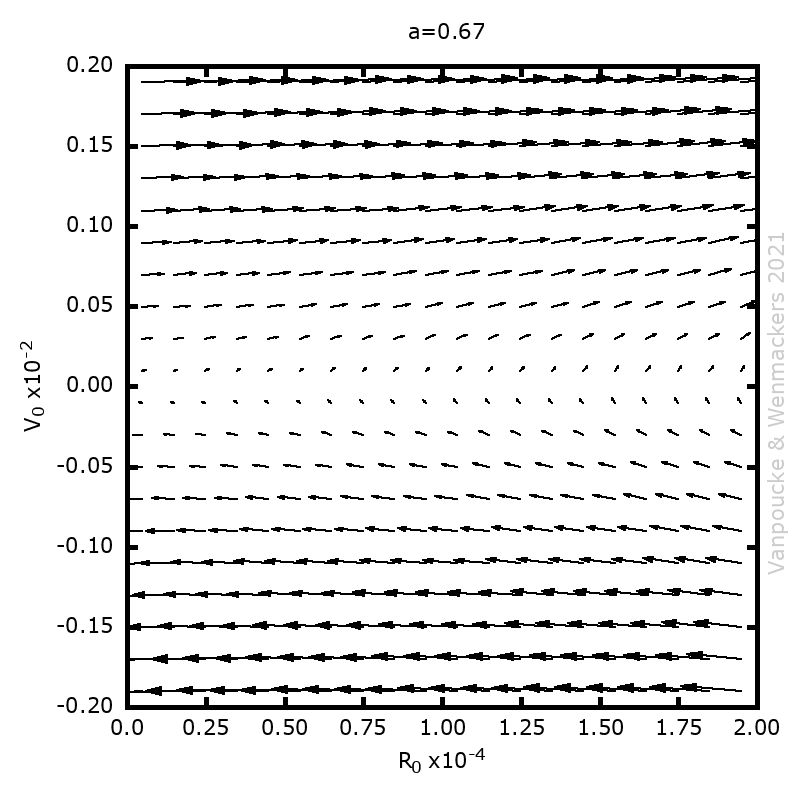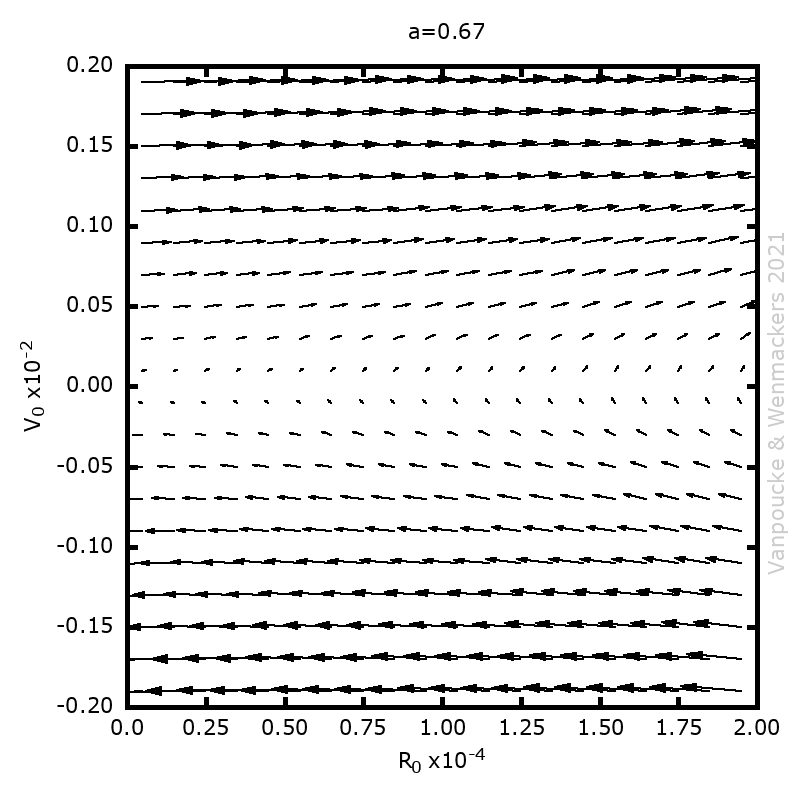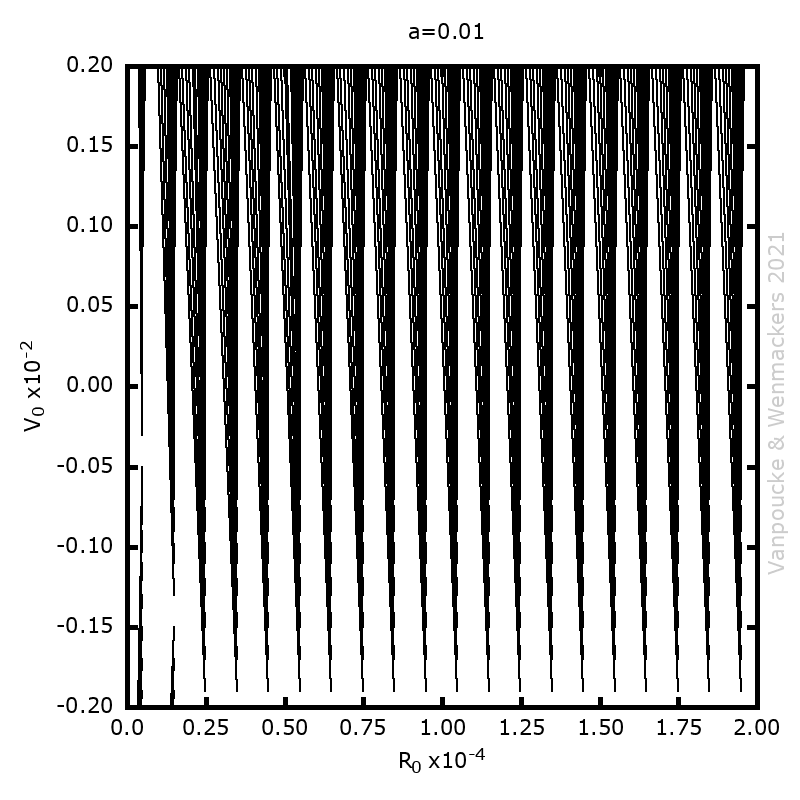
, and }= 0") , with a∈]0; 1[. This example has a physical interpretation as a mass in a uniform gravitational field on a dome of particular shape; the case with a = ½ is known as Norton’s dome. Our approach is based on (1) finite difference equations, which are deterministic; (2) elementary techniques from alpha-theory, a simplified framework for non-standard analysis that allows us to study infinitesimal perturbations; and (3) a uniform prior on the canonical phase space. Our deterministic, hyperfinite grid model allows us to assign probabilities to the solutions of the This allows us to assign probabilities to the solutions of the initial value problem in the original, indeterministic model.
, with a∈]0; 1[. This example has a physical interpretation as a mass in a uniform gravitational field on a dome of particular shape; the case with a = ½ is known as Norton’s dome. Our approach is based on (1) finite difference equations, which are deterministic; (2) elementary techniques from alpha-theory, a simplified framework for non-standard analysis that allows us to study infinitesimal perturbations; and (3) a uniform prior on the canonical phase space. Our deterministic, hyperfinite grid model allows us to assign probabilities to the solutions of the This allows us to assign probabilities to the solutions of the initial value problem in the original, indeterministic model.