

| Authors: | Danny E. P. Vanpoucke and Ken Haenen |
| Journal: | Diam. Relat. Mater 79, 60-69 (2017) |
| doi: | 10.1016/j.diamond.2017.08.009 |
| IF(2017): | 2.232 |
| export: | bibtex |
| pdf: | <DiamRelatMater> |
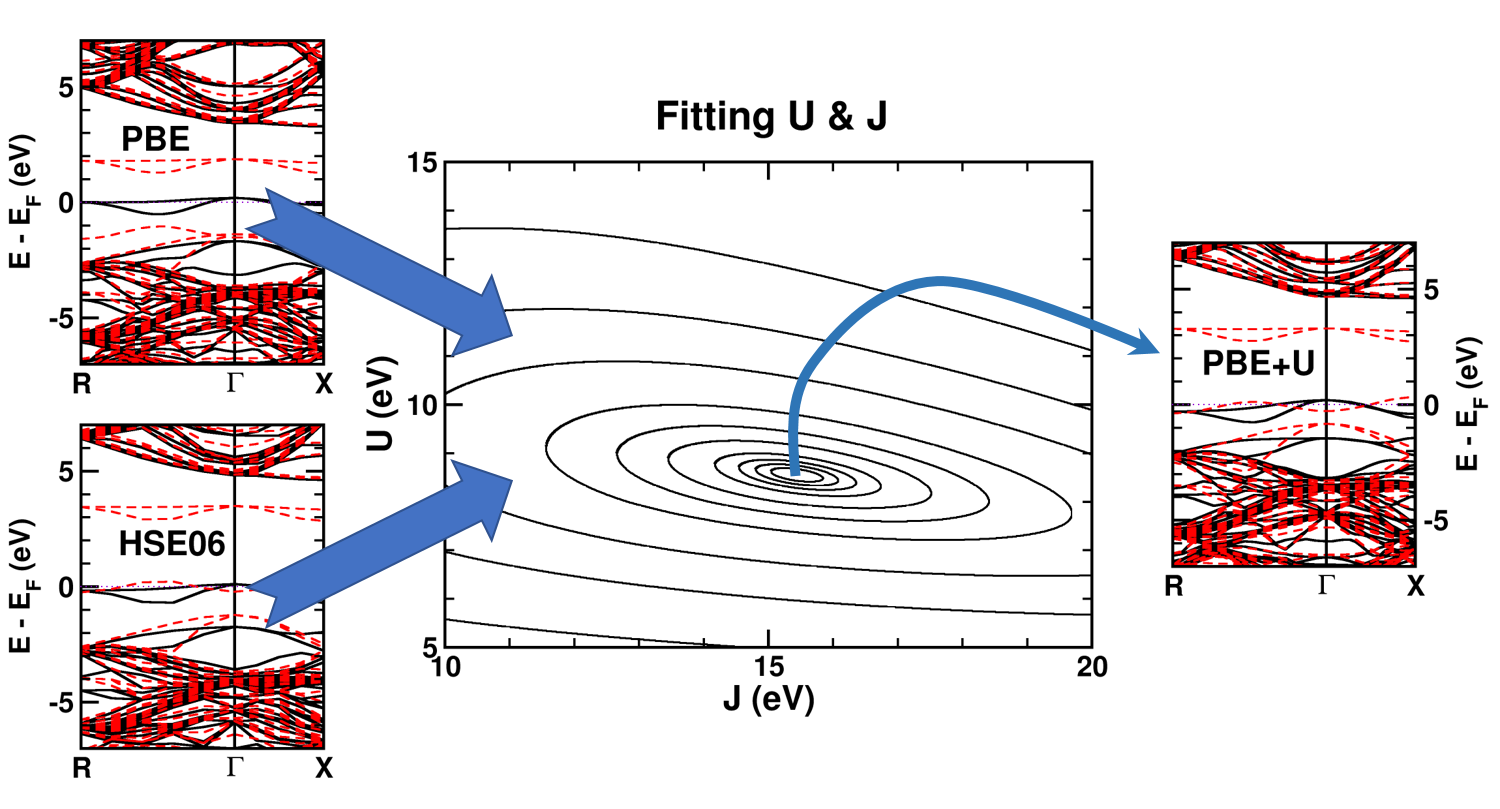 |
| Graphical Abstract: Combining a scan over possible values for U and J with reference electronic structures obtained using the hybrid functional HSE06, DFT+U can be fit to provide hybrid functional quality electronic structures at the cost of DFT calculations. |
Abstract
The neutral C-vacancy is investigated using density functional theory calculations. We show that local functionals, such as PBE, can predict the correct stability order of the different spin states, and that the success of this prediction is related to the accurate description of the local magnetic configuration. Despite the correct prediction of the stability order, the PBE functional still fails predicting the defect states correctly. Introduction of a fraction of exact exchange, as is done in hybrid functionals such as HSE06, remedies this failure, but at a steep computational cost. Since the defect states are strongly localized, the introduction of additional on site Coulomb and exchange interactions, through the DFT+U method, is shown to resolve the failure as well, but at a much lower computational cost. In this work, we present optimized U and J parameters for DFT+U calculations, allowing for the accurate prediction of defect states in defective
diamond. Using the PBE optimized atomic structure and the HSE06 optimized electronic structure as reference, a pair of on-site Coulomb and exchange parameters (U,J) are fitted for DFT+U studies of defects in diamond.
Related:
Poster-presentation: here
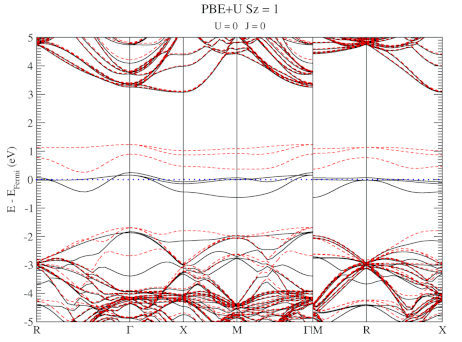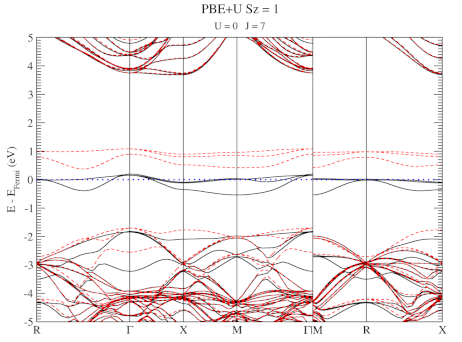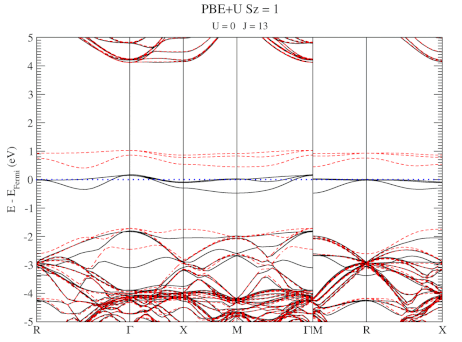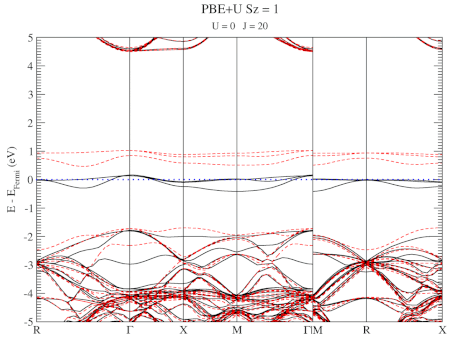
DFT+U series (varying J) for a specific spin state of the C-vacancy defect.