

For a recent project, attempting to investigate Eu dopants in bulk diamond, I ended up simplifying the problem and investigating the C-vacancy in diamond. The setup is simple: take a super cell of diamond, remove 1 carbon atom and calculate. This, however, ended up being a bit more complicated than I had expected.
Removing the single carbon atom gives rise to 4 dangling bonds on the neighboring carbon atoms. The electrons occupying these bonds will gladly interact with one another, giving rise to three different possible spin states:
- All spins oriented the same (Ferromagnetic configuration: ↑↑↑↑ or 4x½ ⇒ Sz=2 spin state)
- Three spins in the same direction and one in the opposite direction ( ↑↑↑↓ or (3x½)-½ ⇒ Sz=1 spin state)
- Two spins up, and two spins down (↑↑↓↓ or (2x½)-(2x½) ⇒Sz=0 spin state)
Starting the calculations without any assumptions gives nice results. Unfortunately they are wrong, and we seem to have ended up in nearby local minima. Including the spin-configurations above as starting assumptions solves the problem, luckily.
The electronic structure is, however, still not a perfect match for experiment. But this is well-known behavior for Density Functional Theory with local functionals such as LDA and PBE. A solution is the use of hybrid functionals (such as HSE06). Conservation of misery kicks in hard at this point, since the latter type of calculations are 1000x as expensive in compute time (and the LDA and PBE calculations aren’t finished in a matter of seconds or minutes, but need several hours on multiple cores). An old methodology to circumvent this problem is the use of Hubbard-U like correction terms (so-called DFT+U). Interestingly for this defect system the two available parameters in a DFT+U setup are independent, and allow for the electronic structure to be perfectly tuned. At the end of the fitting exercise, we now have two additional parameters, which allow us to get electronic structures of hybrid functional quality, but at PBE computational cost.
The evolution of the band-structure as function of the two parameters can be seen below.
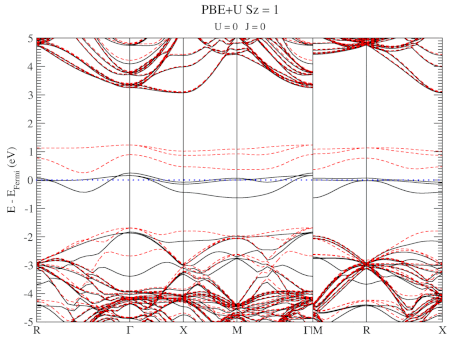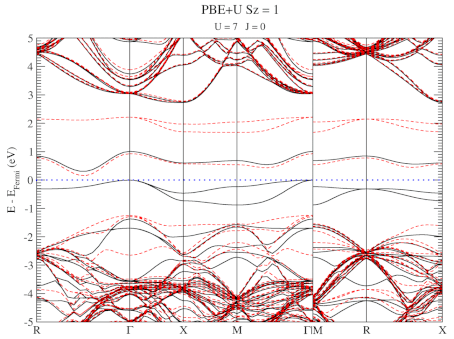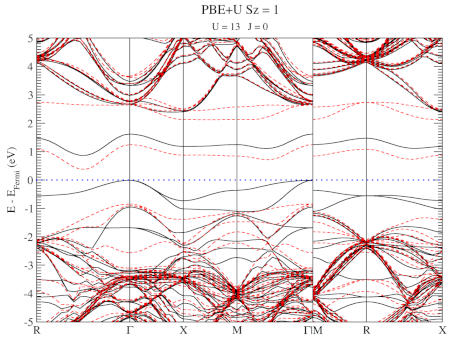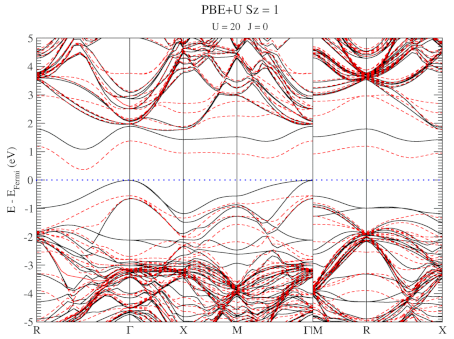
DFT+U series (varying U) for a specific spin state of the C-vacancy defect.
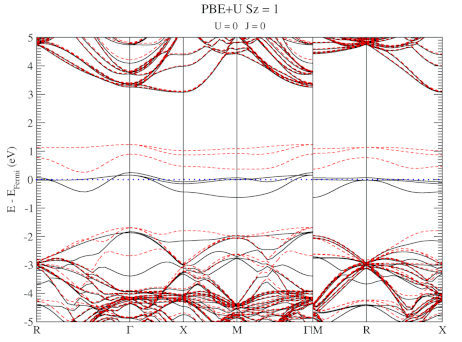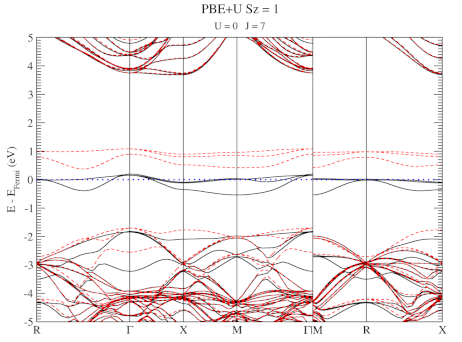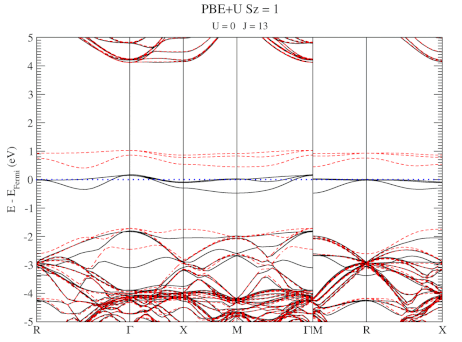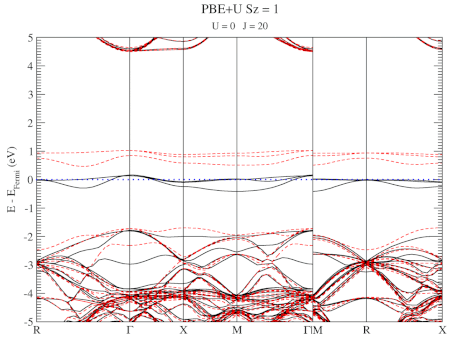
DFT+U series (varying J) for a specific spin state of the C-vacancy defect.