Tag: Diamond and Related Materials
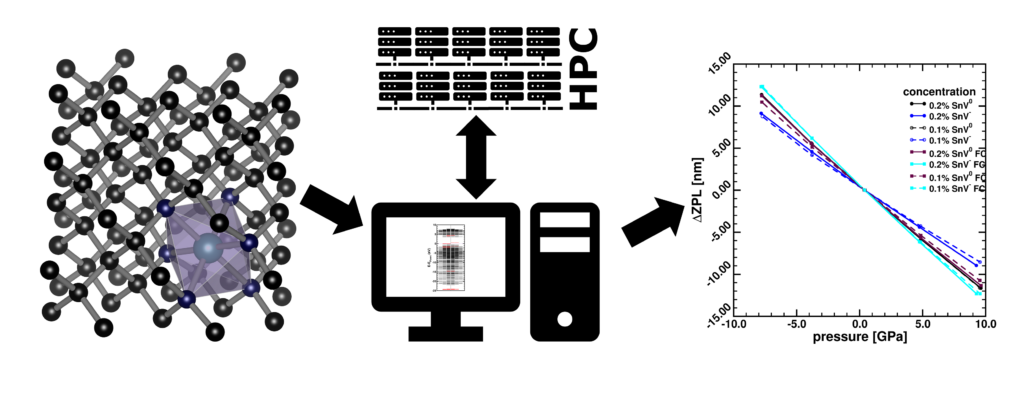 |
| Graphical Abstract: The SnV color center is modelled using first principle calculations to predict the zero-phonon-lines in diamond under hydrostatic strain. |
Among the group-IV vacancy color centers in diamond, the SnV holds promise for photonics based quantum applications. In this work, the Tin-Vacancy (SnV) zero-phonon line (ZPL) and its pressure coefficient are calculated using first principles approaches. The predicted absolute ZPL position is shown to be strongly influenced by the method and supercell size used. The results are therefore extrapolated to the dilute limit allowing for direct comparison with experiments. The importance of identifying the color-center related Kohn–Sham states is highlighted, as well as the shifting of these states due to electron excitations as well as supercell size and k-point position. In contrast to the absolute ZPL positions, the relative position of the SnV0 ZPL is consistently redshifted about 43 nm compared to the SnV– ZPL. In addition, the pressure coefficient is shown to be very robust over different methods, always resulting in a value of about 1.4 nm/GPa, for both SnV0 and SnV–. Finally, the computational accuracy and cost are put into perspective.
Permanent link to this article: https://dannyvanpoucke.be/2026-paper_snvcolorcenter/
-
Filed under blog
-
July 25, 2025
In light of the ever growing interest in AI and ML within the context of materials research, I’m guest editing a special issue together with Konstantin Klyukin from Auburn University. More information can be found on the flyer below.
(And yes the robot and diamond are AI generated, though it took some effort to get it to have the right number or arms, hold a diamond and look sideways at the same time. 😉

Permanent link to this article: https://dannyvanpoucke.be/special-issue-revolutions-in-the-integration-of-artificial-intelligence-and-machine-learning-in-carbon-based-materials-research/
| Authors: |
Kirill N. Boldyrev, Vadim S. Sedov, Danny E.P. Vanpoucke, Victor G. Ralchenko, & Boris N. Mavrin |
| Journal: |
Diam. Relat. Mater 126, 109049 (2022) |
| doi: |
10.1016/j.diamond.2022.109049 |
| IF(2020): |
3.315 |
| export: |
bibtex |
| pdf: |
<DRM> |
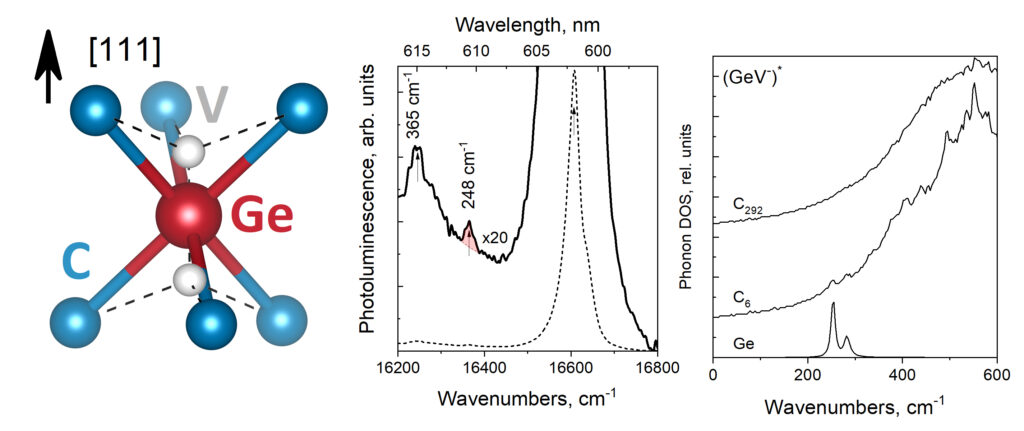 |
| Graphical Abstract: GeV split vacancy defect in diamond and the phonon modes near the ZPL. |
The vibrational behaviour of the germanium-vacancy (GeV) in diamond is studied through its photoluminescence spectrum and first-principles modeled partial phonon density of states. The former is measured in a region below 600 cm−1. The latter is calculated for the GeV center in its neutral, charged, and excited state. The photoluminescence spectrum presents a previously unobserved feature at 248 cm−1 in addition to the well-known peak at 365 cm−1. In our calculations, two localized modes, associated with the GeV center and six nearest carbon atoms (GeC6 cluster) are identified. These correspond to one vibration of the Ge ion along with the [111] orientation of the crystal and one perpendicular to this direction. We propose these modes to be assigned to the two features observed in the photoluminescence spectrum. The dependence of the energies of the localized modes on the GeV-center and their manifestation in experimental optical spectra is discussed.
Permanent link to this article: https://dannyvanpoucke.be/paper_gevpldft_vadim-en/
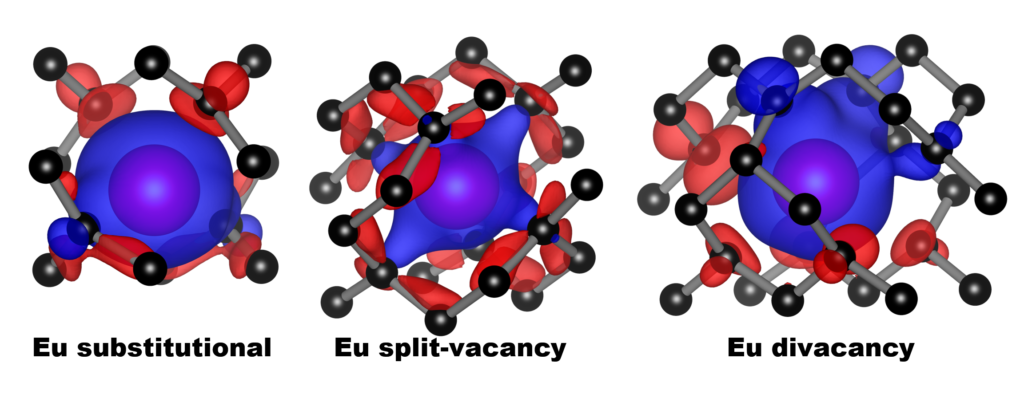 |
| Graphical Abstract: Spin polarization around the various Eu-defect models in diamond. Blue and red represent the up and down spin channels respectively. |
The incorporation of Eu into the diamond lattice is investigated in a combined theoretical-experimental study. The large size of the Eu ion induces a strain on the host lattice, which is minimal for the Eu-vacancy complex. The oxidation state of Eu is calculated to be 3+ for all defect models considered. In contrast, the total charge of the defect-complexes is shown to be negative: -1.5 to -2.3 electron. Hybrid-functional electronic-band-structures show the luminescence of the Eu defect to be strongly dependent on the local defect geometry. The 4-coordinated Eu substitutional dopant is the most promising candidate to present the typical Eu3+ luminescence, while the 6-coordinated Eu-vacancy complex is expected not to present any luminescent behaviour. Preliminary experimental results on the treatment of diamond �films with Eu-containing precursor indicate the possible incorporation of Eu into diamond �films treated by drop-casting. Changes in the PL spectrum, with the main luminescent peak shifting from approximately 614 nm to 611 nm after the growth plasma exposure, and the appearance of a shoulder peak at 625 nm indicate the potential incorporation. Drop-casting treatment with an electronegative polymer material was shown not to be necessary to observe the Eu signature following the plasma exposure, and increased the background
luminescence.
Permanent link to this article: https://dannyvanpoucke.be/paper_eudopingdrmspecial2018-en/
| Authors: |
Danny E. P. Vanpoucke and Ken Haenen |
| Journal: |
Diam. Relat. Mater 79, 60-69 (2017) |
| doi: |
10.1016/j.diamond.2017.08.009 |
| IF(2017): |
2.232 |
| export: |
bibtex |
| pdf: |
<DiamRelatMater> |
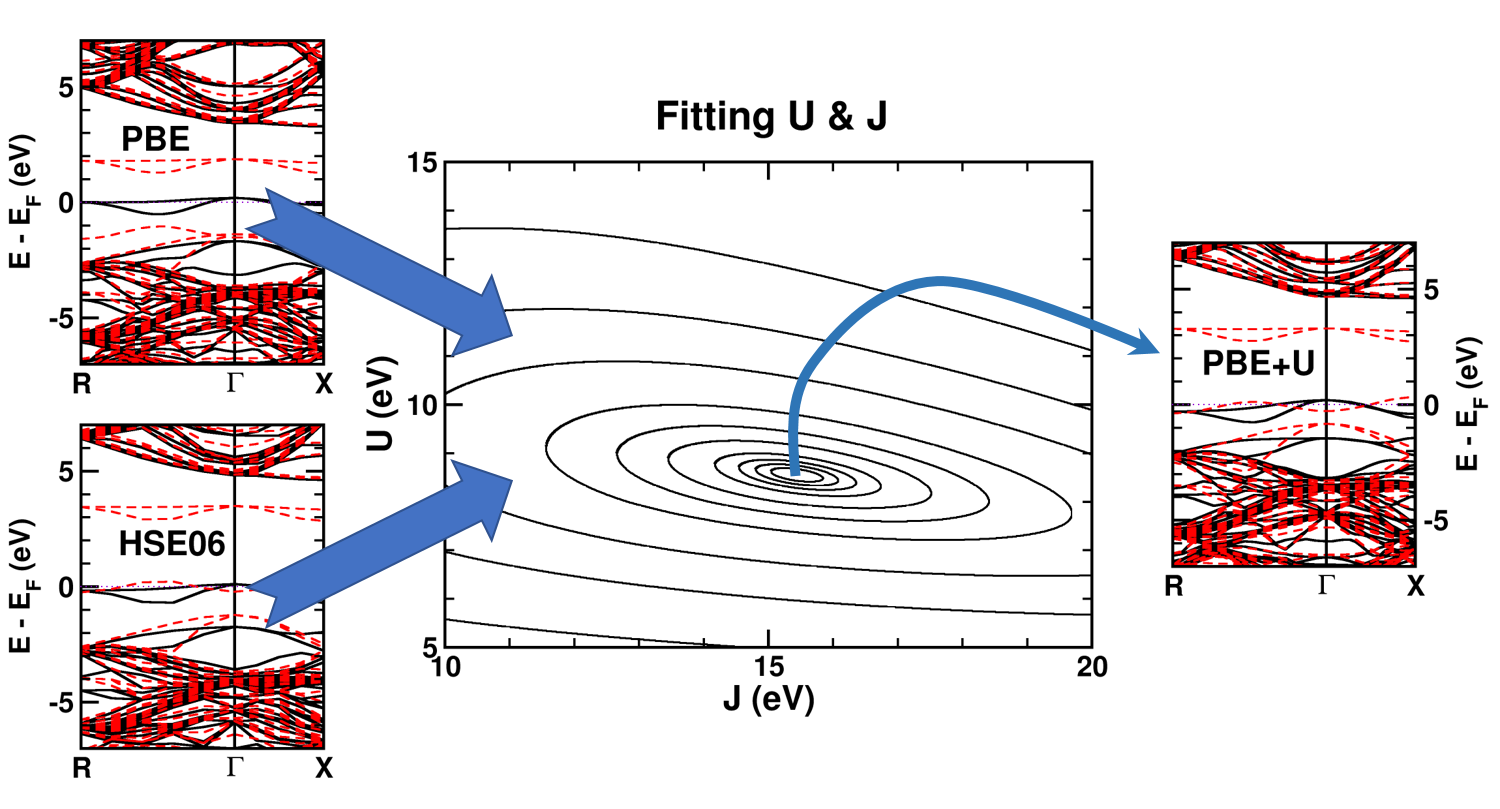 |
| Graphical Abstract: Combining a scan over possible values for U and J with reference electronic structures obtained using the hybrid functional HSE06, DFT+U can be fit to provide hybrid functional quality electronic structures at the cost of DFT calculations. |
The neutral C-vacancy is investigated using density functional theory calculations. We show that local functionals, such as PBE, can predict the correct stability order of the different spin states, and that the success of this prediction is related to the accurate description of the local magnetic configuration. Despite the correct prediction of the stability order, the PBE functional still fails predicting the defect states correctly. Introduction of a fraction of exact exchange, as is done in hybrid functionals such as HSE06, remedies this failure, but at a steep computational cost. Since the defect states are strongly localized, the introduction of additional on site Coulomb and exchange interactions, through the DFT+U method, is shown to resolve the failure as well, but at a much lower computational cost. In this work, we present optimized U and J parameters for DFT+U calculations, allowing for the accurate prediction of defect states in defective
diamond. Using the PBE optimized atomic structure and the HSE06 optimized electronic structure as reference, a pair of on-site Coulomb and exchange parameters (U,J) are fitted for DFT+U studies of defects in diamond.
Poster-presentation: here
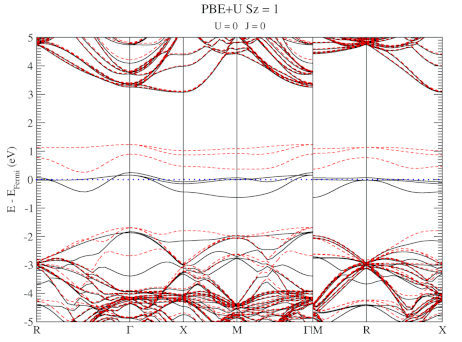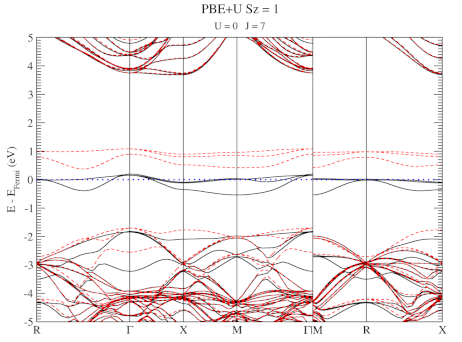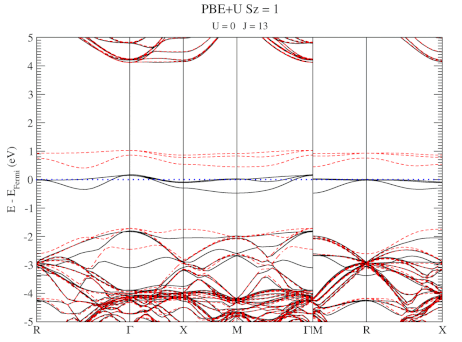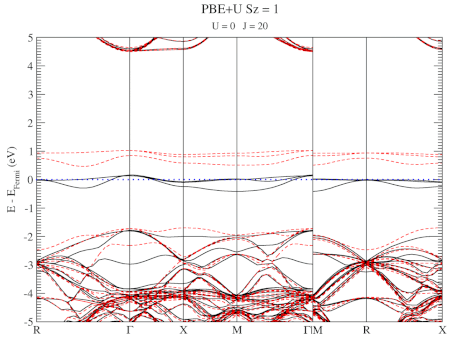
DFT+U series (varying J) for a specific spin state of the C-vacancy defect.
Permanent link to this article: https://dannyvanpoucke.be/paperdrm-diamond-dftu-en/




