After the complexities of week seven, week eight brings the last lecture week of the first quarter of the academic year. After this week, the students of our materiomics program at UHasselt will start studying for a first batch of exams. It also means with this week, their basic courses come to an end and they have all been brought up to speed and to a similar level, needed for the continuation of their study in the materiomics program.
In the bachelor program, the third bachelor chemistry students ended their detailed study of the He atom in the course quantum and computational chemistry with the investigation of its excited states. They learned about the splitting of in singlet and triplet states as well as Fermi-holes and heaps.
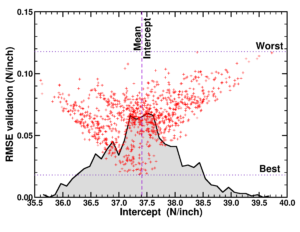
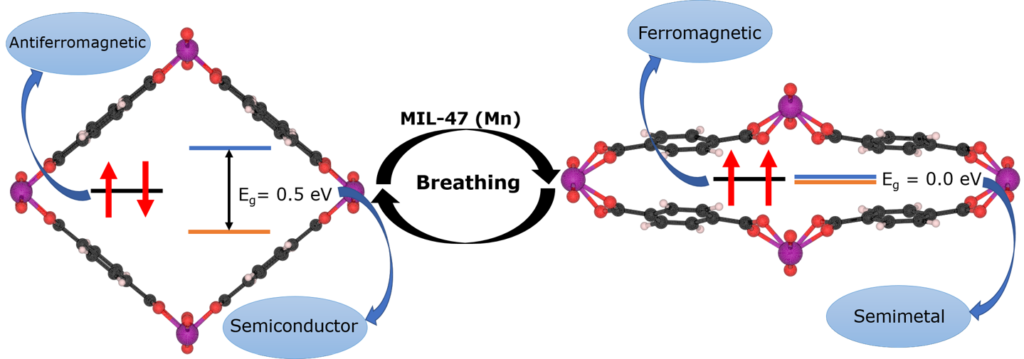
Vulcano-plot of small data model quality of model instances in a large ensemble. Taken from our paper “Small Data Materials Design with Machine Learning: When the Average Model Knows Best“, J. Appl. Phys. 128, 054901 (2020)
The first mater materiomics students got their last lecture in the course Fundamentals of materials modeling, where we looked into some examples of application of machine learning in materials research. We also brought all levels of the course together and imagined how to link these in a multiscale project. Starting from the example of a windmill we discussed the application of computational materials modeling at different scales. For the course Properties of functional materials, the third and final presentation and discussion was held, now focusing on characterization methods. The second master students had response lectures for the courses on Density Functional Theory and Machine learning and artificial intelligence in modern materials science where the various topics of the self study were discussed (e.g., concepts of Neural Networks in case of the latter).
At the end of this week, we have added another 8h of live lectures, putting our semester total at 99h of live lectures. With the workload of the first master materiomics coming to an end, the following chronicles will be biweekly. Upwards and onward to week 9&10.