Category: 2025
| Authors: |
Pieter Verding, Danny E.P. Vanpoucke, Yunus T. Aksoy, Tobias Corthouts, Maria R. Vetrano, and Wim Deferme |
| Journal: |
Adv. Mater. Technol. XX, YY (2025) |
| doi: |
10.1002/admt.202502104 |
| IF(2025): |
6.2 |
| export: |
bibtex |
| pdf: |
<AdvMaterTechnol_XX> |
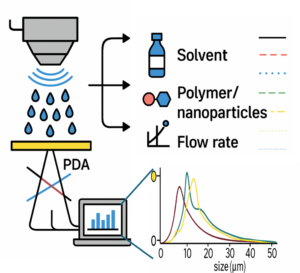 |
| Graphical Abstract: This study explores how machine learning models, trained on small experimental datasets obtained via Phase Doppler Anemometry (PDA), can accurately predict droplet size (D₃₂) in ultrasonic spray coating (USSC). By capturing the influence of ink complexity (solvent, polymer, nanoparticles), power, and flow rate, the model enables precise droplet control paving the way for optimized coatings in advanced functional materials. |
This study examines droplet formation in ultrasonic spray coating (USSC) as a function of ink formulation (solvent, polymer, nanoparticles). First, acetone with polyvinylidene fluoride (PVDF) at concentrations from 0-4.5 wt% is used to examine the effect of polymer additions. Additionally, acetone-based SiO2 nanofluids (0-10 g/L), are explored. Finally, the combination of both polymer (PVDF) and nanoparticles (SiO2) in acetone is studied. Droplet sizes are measured using Phase Doppler Anemometry under varying atomization power and flow rates. Machine Learning (ML) algorithms are employed to develop droplet size models from key spray parameters, including atomization power, flow rate, polymer concentration, and nanoparticle concentration. The model shows significantly higher accuracy than existing empirical models. The model is further validated on IPA-based inks with polyethylenimine (PEIE) or ZnO nanoparticles, and on acetone–cellulose acetate formulations, confirming its robustness across diverse ink systems. In addition to revealing the influence of coating parameters on the droplet formation and distribution, obtained both via experimental validation and ML, this study demonstrates that ML can be effectively applied to small experimental datasets, offering a robust framework for optimizing droplet formation and understanding key spray parameters in USSC for complex, unexplored inks enabling novel coating applications.
Permanent link to this article: https://dannyvanpoucke.be/2025-paper_mldroplets_pieterverding-en/
| Authors: |
Goedele Roos, Danny E.P. Vanpoucke, Ralf Blossey, Marc F. Lensink, and Jane S. Murray |
| Journal: |
J. Chem. Phys. 163, 114112 (2025) |
| doi: |
10.1063/5.0268712 |
| IF(2023): |
3.1 |
| export: |
bibtex |
| pdf: |
<JChemPhys_163> |
 |
| Graphical Abstract: The Electrostatic Potential of water in different situations. On the left two interacting water molecules are shown, while on the right a water molecule interacting with a protein model representation is shown. |
The electrostatic potential plotted on varying contours (VS) of the electron density guides us in the
understanding of how water interactions exactly take place. Water—H2O—is extremely well balanced, having a hydrogen VS,max and an oxygen VS,min of similar magnitude. As such, it has the capacity to donate and accept hydrogen bonds equally well. This has implications for the interactions that water molecules form, which are reviewed here, first in water–small molecule models and then in complex sites as lactose and its crystals and in protein–protein interfaces. Favorable and unfavorable interactions are evaluated from the electrostatic potential plotted on varying contours of the electronic density, allowing these interactions to be readily visualized. As such, with one calculation, all interactions can be analyzed by gradually looking deeper into the electron density envelope and finding the nearly touching contour. Its relation with interaction strength has the electrostatic potential to be used in scoring functions. When properly implemented, we expect this approach to be valuable in modeling and structure validation, avoiding tedious interaction strength calculations. Here, applied to water interactions in a variety of systems, we conclude that all water interactions take the same general form, with water behaving as a “neutral” agent, allowing its interaction partner to determine if it donates or accepts a hydrogen bond, or both, as determined by the highest possible interaction strength(s).
Permanent link to this article: https://dannyvanpoucke.be/2025-paper-wateresp-roos-en/
 |
| Graphical Abstract: Schematic representation of the electrostatic potential within a water molecule along the lines between the atoms. The color background shows the electrostatic potential on the 0.001 a.u. contour of the density. The Vs,min and Vs,max points on the surface are indicated. |
This paper discusses the use of the electrostatic potential in both recent and older literature, with an emphasis upon a 2022 Molecular Physics article by Politzer and Murray entitled “Atoms do exist in molecules: analysis using electrostatic potentials at nuclei“. We discuss electrostatic potentials at nuclei and how they easily lead to atoms in molecules, without physically separating the individual atoms. We further summarize the work by the Politzer group on definitions of atomic radii by means of the electrostatic potential. The earlier studies began in the 1970’s and continued through the 1990’s. Unfortunately, access to these older publications is often limited, cfr. digital libraries often limit the authorized access until a certain publication year, and these papers are often not cited in current publications. Although still being highly interesting and relevant, this older literature is in danger of being lost. Digging into this older literature thus opens up new views. Our feeling is that Peter passed ‘on’ a vision that boundaries do not exist between atoms in molecules, but that some useful and meaningful radii can be obtained using the electrostatic potential between atoms in molecules.
Permanent link to this article: https://dannyvanpoucke.be/2025-paper-esp-politzer-rev-en/
| Authors: |
Thijs G.I. van Wijk, E. Aylin Melan, Rani Mary Joy, Emerick Y. Guillaume, Paulius Pobedinskas, Ken Haenen, and Danny E.P. Vanpoucke |
| Journal: |
Carbon 234, 119928 (2025) |
| doi: |
10.1016/j.carbon.2024.119928 |
| IF(2024): |
10.5 |
| export: |
bibtex |
| pdf: |
<Carbon> |
 |
| Graphical Abstract: Schematic representation of the impact of hydrostatic and linear strain on the Zero Phonon Line of the neutral GeV defect in diamond. |
Color centers in diamond, such as the GeV center, are promising candidates for quantum-based applications. Here, we investigate the impact of strain on the zero-phonon line (ZPL) position of GeV0. Both hydrostatic and linear strain are modeled using density functional theory for GeV0concentrations of 1.61 % down to 0.10 %. We present qualitative and quantitative differences between the two strain types: for hydrostatic tensile and compressive strain, red- and blue-shifted ZPL positions are expected, respectively, with a linear relation between the ZPL shift and the experienced stress. By calculating the ZPL shift for varying GeV0 concentrations, a shift of 0.15 nm/GPa (0.38 meV/GPa) is obtained at experimentally relevant concentrations using a hybrid functional. In contrast, only red-shifted ZPL are found for tensile and compressive linear strain along the ⟨100⟩ direction. The calculated ZPL shift exceeds that of hydrostatic strain by at least one order of magnitude, with a significant difference between tensile and compressive strains: 3.2 and 4.8 nm/GPa (8.1 and 11.7 meV/GPa), respectively. In addition, a peak broadening is expected
due to the lifted degeneracy of the GeV0 eg state, calculated to be about 6 meV/GPa. These calculated results are placed in perspective with experimental observations, showing values of ZPL shifts and splittings of comparable magnitude.
Permanent link to this article: https://dannyvanpoucke.be/2025-paper-strainedgev-en/
| Authors: |
Asif Iqbal Bhatti, Sandeep Kumar, Catharina Jaeken, Michael Sluydts, Danny E.P. Vanpoucke, and Stefaan Cottenier |
| Journal: |
Journal of Materials Chemistry A 13, 526-539 (2025) |
| doi: |
10.1039/D4TA06603K |
| IF(2024): |
10.7 |
| export: |
bibtex |
| pdf: |
<J.Mat.Chem.A> |
 |
| Graphical Abstract: Schematic representation of the LPS material and the variation of results obtained due to slight changes in settings within a High Throughput workflow. |
High-throughput computational screening has become a powerful tool in materials science for identifying promising candidates for specific applications. However, the effectiveness of these methods relies heavily on the accuracy and appropriateness of the underlying models and assumptions. In this study, we use the popular argyrodite solid-state electrolyte family Li6PS5X (X = Cl, Br, I) as a case study to critically examine key steps in high-throughput workflows and highlight potential pitfalls. We demonstrate some of these pitfalls by highlighting the importance of careful structural considerations, including symmetry breaking and site disorder, and examine the difference between 0 K thermodynamic stability and finite-temperature stability based on temperature-dependent Gibbs free energy calculations. Furthermore, we explore the implications of these findings for the ranking of candidate materials in a mini-throughput study in a search space of isovalent analogs to Li6PS5Cl. As a result of these findings, our work underscores the need for balanced trade-offs between computational efficiency and accuracy in high-throughput screenings, and offers guidance for designing more robust workflows that can better bridge the gap between computational predictions and experimental realities.
Permanent link to this article: https://dannyvanpoucke.be/2025-paper-thedevilinthedetails-en/




