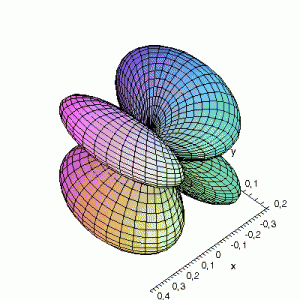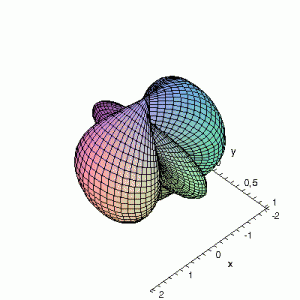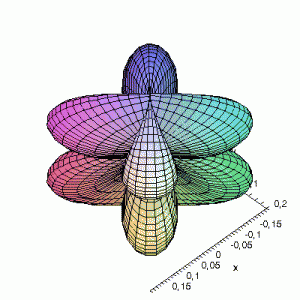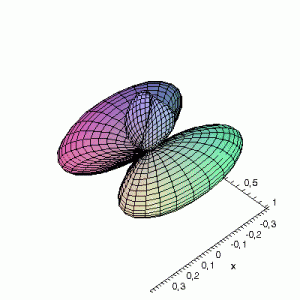Most commented posts
- Phonons: shake those atoms — 3 comments
- Start to Fortran — 1 comment


Oct 28 2014
| Authors: | Danny E. P. Vanpoucke, Patrick Bultinck, Stefaan Cottenier, Veronique Van Speybroeck and Isabel Van Driessche, |
| Journal: | Phys. Rev. B 84, 054110 (2011) |
| doi: | 10.1103/PhysRevB.84.054110 |
| IF(2011): | 3.691 |
| export: | bibtex |
| pdf: | <Phys.Rev.B.> <arXiv> |
The crystal structure of lanthanum cerium oxide (La2Ce2O7) is investigated using ab initio density functional theory calculations. The relative stability of fluorite- and pyrochlore-like structures is studied through comparison of their formation energies. These formation energies show the pyrochlore structure to be favored over the fluorite structure, apparently contradicting the conclusions based on experimental neutron and x-ray diffraction (XRD).
By calculating and comparing XRD spectra for a set of differently ordered and random structures, we show that the pyrochlore structure is consistent with diffraction experiments. For these reasons, we suggest the pyrochlore structure as the ground-state crystal structure for La2Ce2O7.
Oct 28 2014
| Authors: | Danny E. P. Vanpoucke |
| Journal: | Belgian Physical Society Magazine 3, 11-16 (2011) |
| (Featured Article for the BΦ) | |
| pdf: | <local> |
The deposition of small amounts of platinum on a germanium (001) surface gives rise to the formation of monatomic nanowires. These nanowires are defect–and kink-free and their length is only limited by the underlying terrace, to which they are uniquely connected. Using ab initio calculations and simulated scanning tunneling microscopy (STM) images we model these nanowires, and show them to consist of germanium atoms, in contrast to earlier proposed models.
Oct 28 2014
| Authors: | Danny E. P. Vanpoucke and Geert Brocks |
| Journal: | Phys. Rev. B 81, 235434 (2010) |
| doi: | 10.1103/PhysRevB.81.235434 |
| IF(2010): | 3.774 |
| export: | bibtex |
| pdf: | <Phys.Rev.B> <arXiv> |
Using density-functional theory, we investigate the possible adsorption sites of CO molecules on the recently discovered Pt-induced Ge nanowires (NWs) on Ge(001). Calculated scanning tunneling microscope (STM) images are compared to experimental STM images to identify the experimentally observed adsorption sites. The CO molecules are found to adsorb preferably onto the Pt atoms between the Ge nanowire dimer segments. This adsorption site places the CO molecule in between two nanowire dimers, pushing them outward along the NW direction, blocking the nearest equivalent adsorption sites. This explains the observed long-range repulsive interaction between CO molecules on these Pt-induced nanowires.
Oct 28 2014
| Authors: | Danny E. P. Vanpoucke and Geert Brocks |
| Journal: | Phys. Rev. B 81, 085410 (2010) |
| doi: | 10.1103/PhysRevB.81.085410 |
| IF(2010): | 3.774 |
| export: | bibtex |
| pdf: | <Phys.Rev.B> <arXiv> |
We study formation of the nanowires formed after deposition of Pt on a Ge(001) surface. The nanowires form spontaneously after high-temperature annealing. They are thermodynamically stable, only one atom wide and up to a few hundred atoms long. Ab initio density functional theory calculations are performed to identify possible structures of the Pt-Ge(001) surface with nanowires on top. A large number of structures are studied. With nanowires that are formed out of Pt or Ge dimers or mixed Pt-Ge dimers. By comparing simulated scanning tunneling microscopy images (STM) with experimental ones we model the formation of the nanowires and identify the geometries of the different phases in the formation process. We find that the formation of nanowires on a Pt-Ge(001) surface is a complex process based on increasing the Pt density in the top layers of the Ge(001) surface. Most remarkably we find the nanowires to consist of germanium dimers placed in troughs lined by mixed Pt-Ge dimer rows.
Oct 28 2014
| Authors: | Danny E. P. Vanpoucke and Geert Brocks |
| Journal: | Phys. Rev. B 81, 035333 (2010) |
| doi: | 10.1103/PhysRevB.81.035333 |
| IF(2010): | 3.774 |
| export: | bibtex |
| pdf: | <Phys.Rev.B> <arXiv> |
Pt deposited on a Ge(001) surface spontaneously forms nanowire arrays. These nanowires are thermodynamically stable and can be hundreds of atoms long. The nanowires only occur on a reconstructed Pt-Ge-surface where they fill the troughs between the dimer rows on the surface. This unique connection between the nanowires and the underlying substrate make a thorough understanding of the latter necessary for understanding the growth of the nanowires. In this paper we study possible surface reconstructions containing 0.25 and 0.5 of a monolayer of Pt. Comparison of calculated scanning tunneling microscope (STM) images to experimental STM images of the surface reconstruction reveal that the Pt atoms are located in the top layer, creating a structure with rows of alternating Pt-Ge and Ge-Ge dimers in a c(4×2) arrangement. Our results also show that Pt atoms in the second or third layer cannot be responsible for the experimentally observed STM images.
Oct 28 2014
| Authors: | Danny E.P. Vanpoucke |
| Ph.D. Thesis | at University of Twente, The Netherlands |
| date: | September 11th, 2009 |
| Promoters | Prof. Dr. Paul J. Kelly and Dr. Geert H. L. A. Brocks |
| doi: | 10.3990/1.9789036528733 |
| ISBN: | 978-90-365-2873-3 |
| #pages | 193 |
| export: | bibtex |
| pdf: | <PhD.Thesis> <UTwente> |
| research page with more information |
The aim of this thesis: “Ab Initio Study of Pt Induced Nanowires on Ge(001)”, is to model the experimentally observed ‘Pt nanowires’ on Ge(001). These one-atom-thick wires can be hundreds of nanometers long while remaining defect and kink free, providing the ultimate wire any chip designer dreams of. However, experiments show the wires not to be conducting; on the contrary, one-dimensional states are discovered between the wires. To model these nanowires, we combine state of the art density functional calculations with calculated scanning tunneling microscope (STM) images. First, the β-terrace substrate is modeled, showing a checkerboard pattern of Pt-Ge and Ge-Ge surface dimers in a Ge(001)-reconstructed surface.
Starting from this substrate model, different models with increasing Pt density are developed in an iterative fashion showing increasing agreement with the experimentally observed nanowires. We show that, contrary to previous assumptions, the observed wires are not Pt atoms but Ge atoms, explaining the lacking conductivity. The germanium nanowires consist of Ge dimers located in a Pt-lined trough. In addition, the 4×1 periodicity observed in the nanowire-arrays is traced back to the bonds of the Ge nanowire dimers to an extra Pt atom at the bottom of the trough, resulting in the buckling of the nanowires dimers.
In the last part of the thesis we investigate the adsorption of CO on the Ge nanowires under study. The observed adsorption of CO seems to contradict our proposed model due to the high sticking probability of CO on Pt, where it is low on Ge. We show that no contradiction exists. The CO molecules bind to the Pt atoms in the surface, but because they are tilted toward the nanowires, the resulting STM images give the impression that they are located on top of the nanowire giving rise to the apparent contradiction. In this last study, we also discover a very stable CO adsorption configuration in which the CO molecules remain invisible for STM, but could allow for the formation of one-dimensional molecular chains. This would open the door to one-dimensional molecular electronics.

Oct 28 2014
| Authors: | Danny E. P. Vanpoucke and Geert Brocks |
| Book title: | Symposium Z–Computational Nanoscience–How to Exploit Synergy between Predictive Simulations and Experiment |
| proceeding: | Mater. Res. Soc. Symp. Proc. 1177, 1177-Z03-09 (2009) |
| doi: | 10.1557/PROC-1177-Z03-09 |
| export: | bibtex |
| pdf: | <MRS Proceeding> <arXiv> |
Nanowire (NW) arrays form spontaneously after high temperature annealing of a sub monolayer deposition of Pt on a Ge(001) surface. These NWs are a single atom wide, with a length limited only by the underlying beta-terrace to which they are uniquely connected. Using ab-initio density functional theory (DFT) calculations we study possible geometries of the NWs and substrate. Direct comparison to experiment is made via calculated scanning tunneling microscope (STM) images. Based on these images, geometries for the beta-terrace and the NWs are identified, and a formation path for the nanowires as function of increasing local Pt density is presented. We show the beta-terrace to be a dimer row surface reconstruction with a checkerboard pattern of Ge-Ge and Pt-Ge dimers. Most remarkably, comparison of calculated to experimental STM images shows the NWs to consist of germanium atoms embedded in the Pt-lined troughs of the underlying surface, contrary to what was assumed previously in experiments.
Oct 28 2014
| Authors: | Danny E. P. Vanpoucke and Geert Brocks |
| Journal: | Phys. Rev. B 77, 241308 (2008) |
| doi: | 10.1103/PhysRevB.77.241308 |
| IF(2008): | 3.322 |
| export: | bibtex |
| pdf: | <Phys.Rev.B> <arXiv> <UTwentePublications> |
Pt deposited onto a Ge(001) surface gives rise to the spontaneous formation of atomic nanowires on a mixed Pt-Ge surface after high-temperature annealing. We study possible structures of the mixed surface and the nanowires by total energy density functional theory calculations. Experimental scanning-tunneling microscopy images are compared to the calculated local densities of states. On the basis of this comparison and the stability of the structures, we conclude that the formation of nanowires is driven by an increased concentration of Pt atoms in the Ge surface layers. Surprisingly, the atomic nanowires consist of Ge instead of Pt atoms.
Jan 01 2010
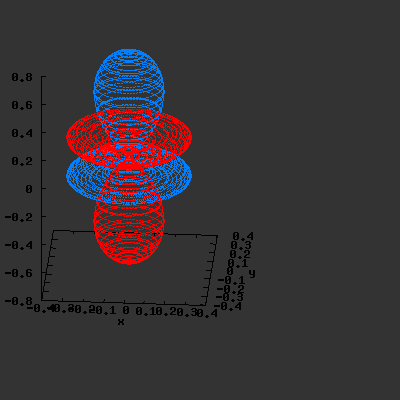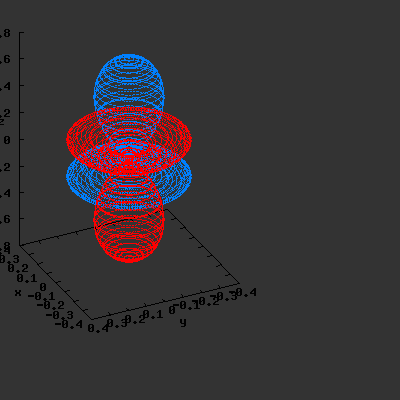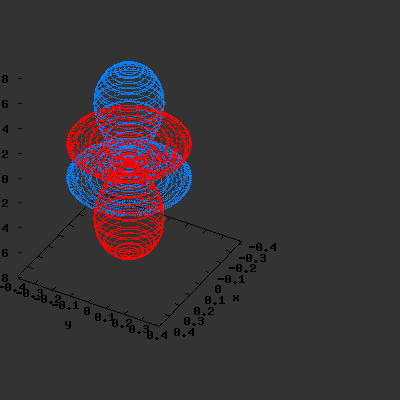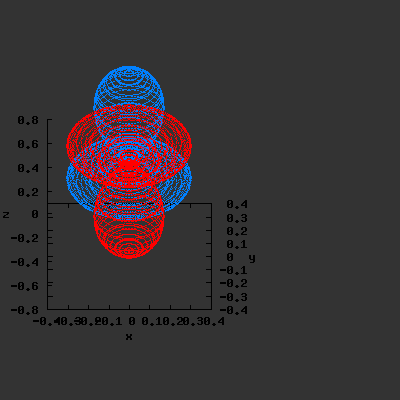
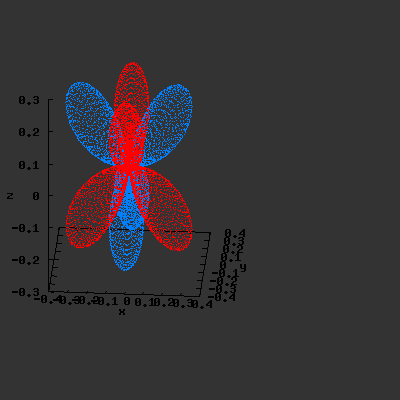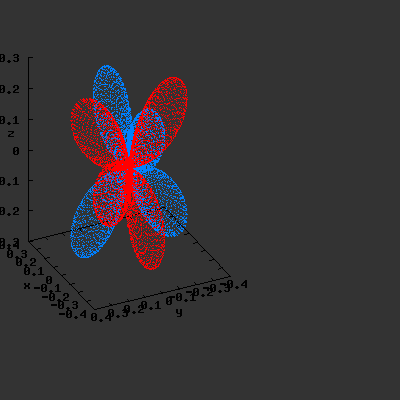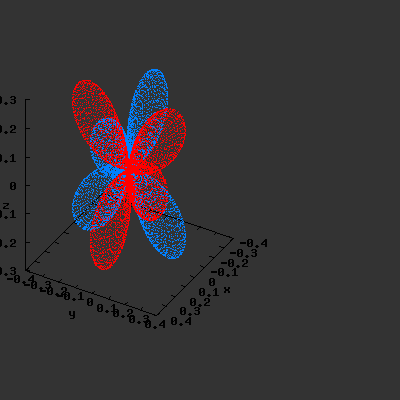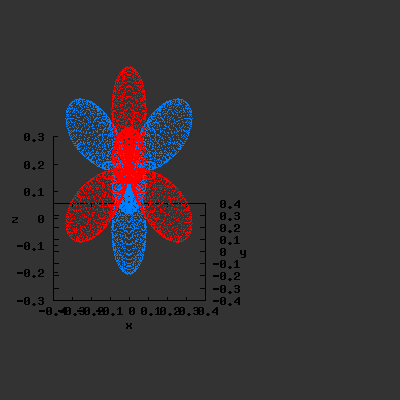
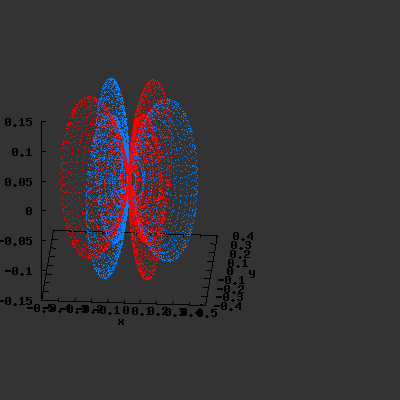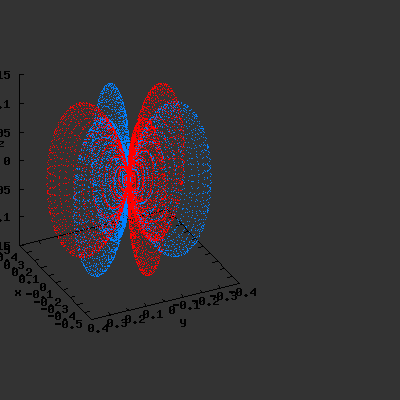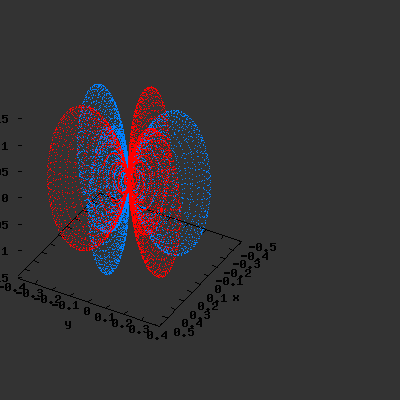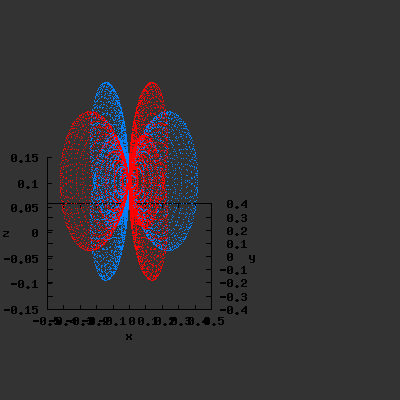
3D gnuplot-gif-animations of the f-orbitals S03(θ,φ), S23(θ,φ) and
S33(θ,φ). In the images presented, the blue part represents the positive phase, and the red part the negative phase. Note that in gnuplot, the spherical coordinate θ is defined as π/2 – θ. Other than that the definitions of φ and θ coincide with those used in Griffiths’ Introduction to Quantum Mechanics.
For those interested: animations in gnuplot are only available for gnuplot versions > 4.0 (which at the moment of making these animations, was still in beta version).
Jan 01 2000
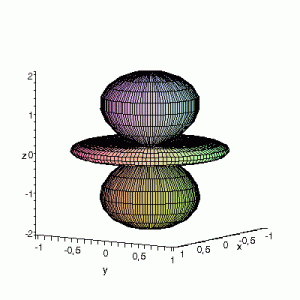
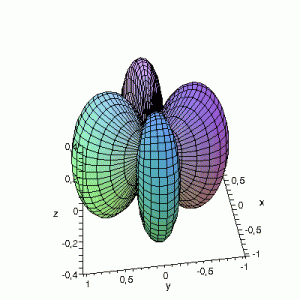
3D Maple-images of the d-orbitals S02(θ,φ), S12(θ,φ) en S22(θ,φ). Note that the spherical coordinates (θ and φ) used by Maple are reversed compared to the definitions used in Griffiths’ Introduction to Quantum Mechanics (the latter being the more standard definition in physics and mathematics courses).