| Authors: | Emerick Y. Guillaum, Danny E. P. Vanpoucke, Rozita Rouzbahani, Luna Pratali Maffei, Matteo Pelucchi, Yoann Olivier, Luc Henrard, & Ken Haenen |
| Journal: | Carbon 222, 118949 (2024) |
| doi: | 10.1016/j.carbon.2024.118949 |
| IF(2022): | 10.9 |
| export: | bibtex |
| pdf: | <Carbon> |
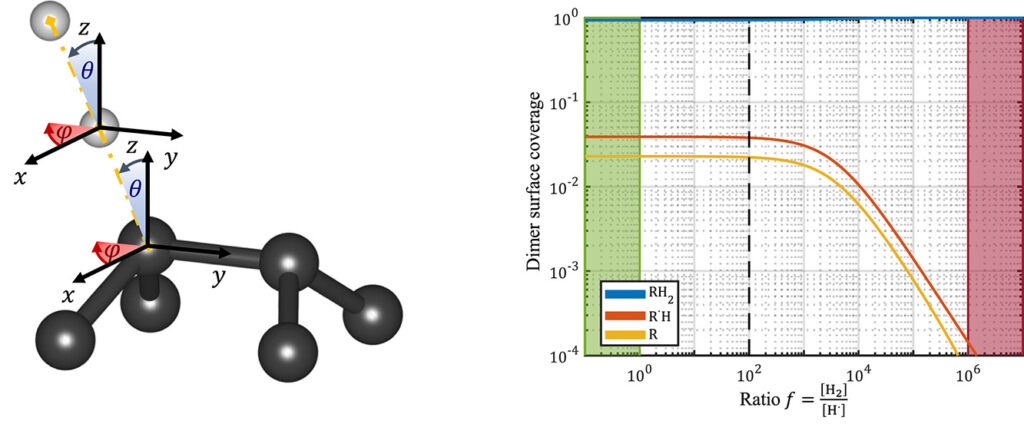 |
| Graphical Abstract: (left) Ball-and-stick representation of aH adsorption/desorption reaction mediated through a H radical. (right) Monte Carlo estimates of the H coverage of the diamond surface at different temperatures based on quantum mechanically determined reaction barriers and reaction rates. |
Abstract
Hydrogen radical attacks and subsequent hydrogen migrations are considered to play an important role in the atomic-scale mechanisms of diamond chemical vapour deposition growth. We perform a comprehensive analysis of the reactions involving H-radical and vacancies on H-passivated diamond surfaces exposed to hydrogen radical-rich atmosphere. By means of first principles calculations—density functional theory and climbing image nudged elastic band method—transition states related to these mechanisms are identified and characterised. In addition, accurate reaction rates are computed using variational transition state theory. Together, these methods provide—for a broad range of temperatures and hydrogen radical concentrations—a picture of the relative likelihood of the migration or radical attack processes, along with a statistical description of the hydrogen coverage fraction of the (100) H-passivated surface, refining earlier results via a more thorough analysis of the processes at stake. Additionally, the migration of H-vacancy is shown to be anisotropic, and occurring preferentially across the dimer rows of the reconstructed surface. The approach used in this work can be generalised to other crystallographic orientations of diamond surfaces or other semiconductors.