Category: publications
| Authors: |
Seyyed Amin Rounaghi, Danny E.P. Vanpoucke, Hossein Eshghi, Sergio Scudino, Elaheh Esmaeili, Steffen Oswald and Jürgen Eckert |
| Journal: |
J. Alloys Compd. 729, 240-248 (2017) |
| doi: |
10.1016/j.jallcom.2017.09.168 |
| IF(2017): |
3.779 |
| export: |
bibtex |
| pdf: |
<J.Alloys Compd.> |
 |
| Graphical Abstract: Evolution of the end products as function of Al and N content during ball-milling synthesis of AlN. |
A versatile ball milling process was employed for the synthesis of hexagonal aluminum nitride (h-AlN) through the reaction of metallic aluminum with melamine. A combined experimental and theoretical study was carried out to evaluate the synthesized products. Milling intermediates and products were fully characterized via various techniques including XRD, FTIR, XPS, Raman and TEM. Moreover, a Boltzmann distribution model was proposed to investigate the effect of milling energy and reactant ratios on the thermodynamic stability and the proportion of different milling products. According to the results, the reaction mechanism and milling products were significantly influenced by the reactant ratio. The optimized condition for AlN synthesis was found to be at Al/M molar ratio of 6, where the final products were consisted of nanostructured AlN with average crystallite size of 11 nm and non-crystalline heterogeneous carbon.
Permanent link to this article: https://dannyvanpoucke.be/paperdoped-nitrides-iii-en/
| Authors: |
Seyyed Amin Rounaghi, Danny E.P. Vanpoucke, Hossein Eshghi, Sergio Scudino, Elaheh Esmaeili, Steffen Oswald and Jürgen Eckert |
| Journal: |
Phys. Chem. Chem. Phys. 19, 12414-12424 (2017) |
| doi: |
10.1039/C7CP00998D |
| IF(2017): |
3.906 |
| export: |
bibtex |
| pdf: |
<Phys.Chem.Chem.Phys.> |
Nowadays, the development of highly efficient routes for the low cost synthesis of nitrides is greatly growing. Mechanochemical synthesis is one of those promising techniques which is conventionally employed for the synthesis of nitrides by long term milling of metallic elements under pressurized N2 or NH3 atmosphere (A. Calka and J. I. Nikolov, Nanostruct. Mater., 1995, 6, 409-412). In the present study, we describe a versatile, room-temperature and low cost mechanochemical process for the synthesis of nanostructured metal nitrides (MNs), carbonitrides (MCNs) and carbon nitride (CNx). Based on this technique, melamine as a solid nitrogen-containing organic compound (SNCOC) is ball milled with four different metal powders (Al, Ti, Cr and V) to produce nanostructured AlN, TiCxN1-x, CrCxN1-x, and VCxN1-x (x~0.05). Both theoretical and experimental techniques are implemented to determine the reaction intermediates, products, by-products and finally, the mechanism underling this synthetic route. According to the results, melamine is polymerized in the presence of metallic elements at intermediate stages of the milling process, leading to the formation of a carbon nitride network. The CNx phase subsequently reacts with the metallic precursors to form MN, MCN or even MCN-CNx nano-composites depending on the defect formation energy and thermodynamic stability of the corresponding metal nitride, carbide and C/N co-doped structures.
Permanent link to this article: https://dannyvanpoucke.be/doped-nitrides-ii-en/
| Authors: |
Danny E. P. Vanpoucke |
| Journal: |
J. Phys. Chem. C 121(14), 8014-8022 (2017) |
| doi: |
10.1021/acs.jpcc.7b01491 |
| IF(2017): |
4.484 |
| export: |
bibtex |
| pdf: |
<J.Phys.Chem.C> |
 upon OH-functionalization of the BDC linker.") |
| Graphical Abstract: Evolution of the electronic band structure of MIL-47(V) upon OH-functionalization of the BDC linker. The π-orbital of the BDC linker splits upon functionalisation, and the split-off π-band moves up into the band gap, effectively reducing the latter. |
Metal–organic frameworks (MOFs) have gained much interest due to their intrinsic tunable nature. In this work, we study how linker functionalization modifies the electronic structure of the host MOF, more specifically, the MIL-47(V)-R (R = −F, −Cl, −Br, −OH, −CH3, −CF3, and −OCH3). It is shown that the presence of a functional group leads to a splitting of the π orbital on the linker. Moreover, the upward shift of the split-off π-band correlates well with the electron-withdrawing/donating nature of the functional groups. For halide functional groups the presence of lone-pair back-donation is corroborated by calculated Hirshfeld-I charges. In the case of the ferromagnetic configuration of the host MIL-47(V+IV) material a half-metal to insulator transition is noted for the −Br, −OCH3, and −OH functional groups, while for the antiferromagnetic configuration only the hydroxy group results in an effective reduction of the band gap.
Permanent link to this article: https://dannyvanpoucke.be/mof-mil47-linkerfunct-en/
| Authors: |
S. A. Rounaghi, H. Eshghi, S. Scudino, A. Vyalikh, D. E. P. Vanpoucke, W. Gruner,
S. Oswald, A. R. Kiani Rashid, M. Samadi Khoshkhoo, U. Scheler and J. Eckert |
| Journal: |
Scientific Reports 6, 33375 (2016) |
| doi: |
10.1038/srep33375 |
| IF(2016): |
4.259 |
| export: |
bibtex |
| pdf: |
<Sci.Rep.> (open access) |
Hexagonal Aluminium nitride (h-AlN) is an important wide-bandgap semiconductor material which is conventionally fabricated by high temperature carbothermal reduction of alumina under toxic ammonia atmosphere. Here we report a simple, low cost and potentially scalable mechanochemical procedure for the green synthesis of nanostructured h-AlN from a powder mixture of Aluminium and melamine precursors. A combination of experimental and theoretical techniques has been employed to provide comprehensive mechanistic insights on the reactivity of melamine, solid state metalorganic interactions and the structural transformation of Al to h-AlN under non-equilibrium ball milling conditions. The results reveal that melamine is adsorbed through the amine groups on the Aluminium surface due to the long-range van der Waals forces. The high energy provided by milling leads to the deammoniation of melamine at the initial stages followed by the polymerization and formation of a carbon nitride network, by the decomposition of the amine groups and, finally, by the subsequent diffusion of nitrogen into the Aluminium structure to form h-AlN
Permanent link to this article: https://dannyvanpoucke.be/paper2016_scirepalnitride-en/
| Authors: |
Arthur De Vos, Kurt Lejaeghere, Danny E. P. Vanpoucke, Jonas J. Joos, Philippe F. Smet, and Karen Hemelsoet |
| Journal: |
Inorg. Chem. 55(5), 2402-2412 (2016) |
| doi: |
10.1021/acs.inorgchem.5b02805 |
| IF(2016): |
4.857 |
| export: |
bibtex |
| pdf: |
<Inorg.Chem> |
 Ball-and-stick model of zinc gallate (right) density of states of Cr doped zinc gallate.") |
| Graphical Abstract: First-principles simulations on zinc gallate solid phosphors (ZGO) containing a chromium dopant and antisite defects (left) rationalize the attractive interactions between the various elements. A large number of antisite pair configurations is investigated and compared with isolated antisite defects. Defect energies point out the stability of the antisite defects in ZGO. Local structural distortions are reported, and charge transfer mechanisms are analyzed based on theoretical density of states (right) and Hirshfeld-I charges. |
Zinc gallate doped with chromium is a recently developed near-infrared emitting persistent phosphor, which is now extensively studied for in vivo bioimaging and security applications. The precise mechanism of this persistent luminescence relies on defects, in particular, on antisite defects and antisite pairs. A theoretical model combining the solid host, the dopant, and/or antisite defects is constructed to elucidate the mutual interactions in these complex materials. Energies of formation as well as dopant, and defect energies are calculated through density-functional theory simulations of large periodic supercells. The calculations support the chromium substitution on the slightly distorted octahedrally coordinated gallium site, and additional energy levels are introduced in the band gap of the host. Antisite pairs are found to be energetically favored over isolated antisites due to significant charge compensation as shown by calculated Hirshfeld-I charges. Significant structural distortions are found around all antisite defects. The local Cr surrounding is mainly distorted due to a ZnGa antisite. The stability analysis reveals that the distance between both antisites dominates the overall stability picture of the material containing the Cr dopant and an antisite pair. The findings are further rationalized using calculated densities of states and Hirshfeld-I charges.
Permanent link to this article: https://dannyvanpoucke.be/paper2016_inorgchemzgodoping-en/
| Authors: |
Danny E. P. Vanpoucke, |
| Journal: |
Developments in Strategic Ceramic Materials:
Ceramic Engineering and Science Proceedings 36(8), 323-334 (2016)
(ICACC 2015 conference proceeding) |
| Editors: |
Waltraud M. Kriven, Jingyang Wang, Dongming Zhu,Thomas Fischer, Soshu Kirihara |
| ISBN: |
978-1-119-21173-0 |
| webpage: |
Wiley-VCH |
| export: |
bibtex |
| pdf: |
<preprint> |
In contemporary materials research, we are able to create and manipulate materials at ever smaller scales: the growth of wires with nanoscale dimensions and the deposition of layers with a thickness of only a few atoms are just two examples that have become common practice. At this small scale, quantum mechanical effects become important, and this is where computational materials research comes into play. Using clever approximations, it is possible to simulate systems with a scale relevant for experiments. The resulting theoretical models provide fundamental insights in the underlying physics and chemistry, essential for advancing modern materials research. As a result, the use of computational experiments is rapidly becoming an important tool in materials research both for predictive modeling of new materials and for gaining fundamental insights in the behavior of existing materials. Computer and lab experiments have complementary limitations and strengths; only by combining them can the deepest fundamental secrets of a material be revealed.
In this paper, we discuss the application of computational materials science for nanowires on semiconductor surfaces, ceramic materials and flexible metal-organic frameworks, and how direct comparison can advance insight in the structure and properties of these materials.
Permanent link to this article: https://dannyvanpoucke.be/paper2016_icacc_gyif-en/
| Authors: |
Danny E. P. Vanpoucke, |
| Journal: |
Developments in Strategic Ceramic Materials:
Ceramic Engineering and Science Proceedings 36(8), 169-177 (2016)
(ICACC 2015 conference proceeding) |
| Editors: |
Waltraud M. Kriven, Jingyang Wang, Dongming Zhu,Thomas Fischer, Soshu Kirihara |
| ISBN: |
978-1-119-21173-0 |
| webpage: |
Wiley-VCH |
| export: |
bibtex |
| pdf: |
<preprint> |
In layered ceramic superconductor architectures, CeO2 buffer layers are known to form micro cracks during the fabrication process. To prevent this crack formation, doping of the CeO2 layer has been suggested. In this theoretical study, the influence of dopants (both tetravalent and aliovalent) on the mechanical and structural properties of CeO2 is investigated by means of density functional theory. Group IVa and IVb dopants show clearly distinct stability, with the former favouring interface and surface doping, while the latter favour uniform bulk doping. This behaviour is linked to the dopant electronic structure. The presence of charge compensating vacancies is shown to complicate the mechanical and structural picture for aliovalent dopants. We find that the vacancies often counteract the dopant modifications of the host material. In contrast, all dopants show an inverse relation between the bulk modulus and thermal expansion coefficient, independent of their valency and the presence of oxygen vacancies. Based on the study of these idealized systems, new dopants are suggested for applications.
Permanent link to this article: https://dannyvanpoucke.be/paper2016_icacc_virtmater-en/
| Authors: |
Bart Bueken, Frederik Vermoortele, Danny E. P. Vanpoucke, Helge Reinsch, Chih-Chin Tsou, Pieterjan Valvekens, Trees De Baerdemaeker, Rob Ameloot, Christine E. A. Kirschhock, Veronique Van Speybroeck, James M. Mayer and Dirk De Vos |
| Journal: |
Angew. Chem. Int. Ed. 54(47), 13912-13917 (2015) |
| doi: |
10.1002/anie.201505512 |
| IF(2015): |
11.705 |
| export: |
bibtex |
| pdf: |
<Angew.Chem.Int.Ed.> |
The synthesis of titanium-carboxylate metal-organic frameworks (MOFs) is hampered by the high reactivity of the commonly employed alkoxide precursors. Here, we present an innovative approach to Ti-based MOFs using titanocene dichloride to synthesize COK-69, the first breathing Ti-MOF built up of trans-1,4- cyclohexanedicarboxylate linkers and an unprecedented [TiIV3(µ3-O)(O)2(COO)6] cluster. The photoactive properties of COK-69 were investigated in-depth by proton-coupled electron transfer experiments, which revealed that up to one TiIV per cluster can be photoreduced to TiIII, while preserving the structural integrity of the framework. From molecular modeling, the electronic structure of COK-69 was determined and a band gap of 3.77 eV was found.
Permanent link to this article: https://dannyvanpoucke.be/paper2015_aniecok69-en/
-
Filed under Cover
-
November 13, 2015
| Authors: |
Thomas Bogaerts, Louis Vanduyfhuys, Danny E.P. Vanpoucke, Jelle Wieme, Michel Waroquier, Pascal Van Der Voort, and Veronique Van Speybroeck, |
| Journal: |
CrystEngComm. 17(45), 8565 (2015) |
| doi: |
10.1039/C5CE90198G |
| IF(2015): |
3.849 |
| export: |
bibtex |
| pdf: |
<CrystEngComm> |
The cover image depicts an X-ray beam hitting a sample of MIL-47(V) Metal-Organic Framework (reddish powder), resulting in an X-ray diffraction pattern. This leads to the atomic structure on the left, Where the spin-densities are indicated for the anti-ferromagnetic ground state. (The related paper can be found here.)

Permanent link to this article: https://dannyvanpoucke.be/paper2015_xrdthomasdannycover-en/
| Authors: |
Kevin Hendrickx, Danny E.P. Vanpoucke, Karen Leus, Kurt Lejaeghere,
Andy Van Yperen-De Deyne, Veronique Van Speybroeck, Pascal Van Der
Voort, and Karen Hemelsoet |
| Journal: |
Inorg. Chem. 54(22), 10701-10710 (2015) |
| doi: |
10.1021/acs.inorgchem.5b01593 |
| IF(2015): |
4.820 |
| export: |
bibtex |
| pdf: |
<Inorg.Chem.> |
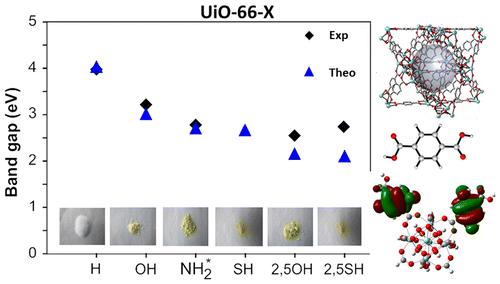
Linker-functionalization of UiO-66 modifies the optical band gap and thus the color of the MOF.
A combined theoretical and experimental study is performed in order to elucidate the eff�ects of linker functional groups on the photoabsorption properties of UiO-66-type materials. This study, in which both mono- and di-functionalized linkers (with X= -OH, -NH2, -SH) are studied, aims to obtain a more complete picture on the choice of functionalization. Static Time-Dependent Density Functional Theory (TD-DFT) calculations combined with Molecular Dynamics simulations are performed on the linkers and compared to experimental UV/VIS spectra, in order to understand the electronic eff�ects governing the absorption spectra. Di-substituted linkers show larger shifts compared to mono-substituted variants, making them promising candidates for further study as photocatalysts. Next, the interaction between the linker and the inorganic part of the framework is theoretically investigated using a cluster model. The proposed Ligand-to-Metal-Charge Transfer (LMCT) is theoretically observed and is influenced by the differences in functionalization. Finally, computed electronic properties of the periodic UiO-66 materials reveal that the band gap can be altered by linker functionalization and ranges from 4.0 down to 2.2 eV. Study of the periodic Density of States (DOS) allows to explain the band gap modulations of the framework in terms of a functionalization-induced band in the band gap of the original UiO-66 host.
Permanent link to this article: https://dannyvanpoucke.be/paper2015_inorgchemuio66-en/

