Happy New Year
2020 will forever be the year of viruses for me and a lot of us. At Maastricht University, the year started with a university wide cyber-attack with ransomware. After the computer-viruses came the human viruses, with COVID-19 shutting down one country after the other, and shutting down education systems as well.
Hopefully 2021 will be better behaved, though we know already some of the hurdles which will make life interesting the coming year. COVID-19 is far from over, and it will take at least a year to vaccinate everyone. Furthermore, as of the first of today, the United Kingdom is no longer a part of the EU, making travel inside Europe a little harder again.
But before we launch into these new and interesting times, lets look back at 2020 one last time, keeping up with tradition. What have I done during the last year of academic merit.
1. Publications: +6 (and currently a handful in progress)
-
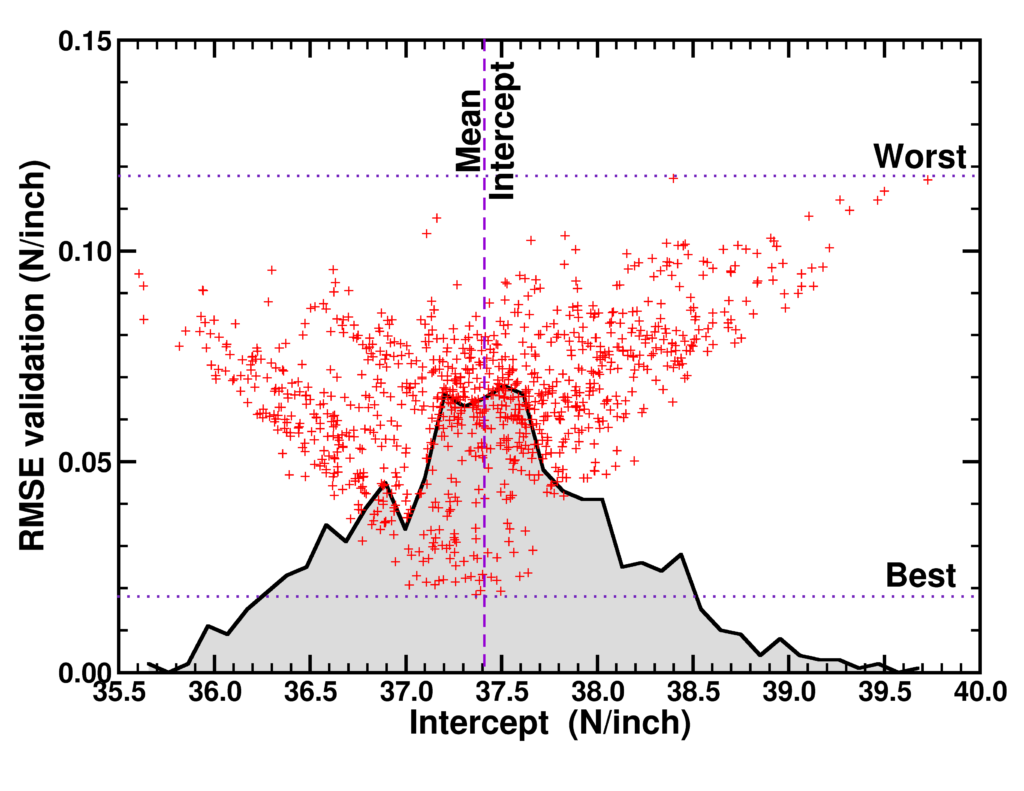
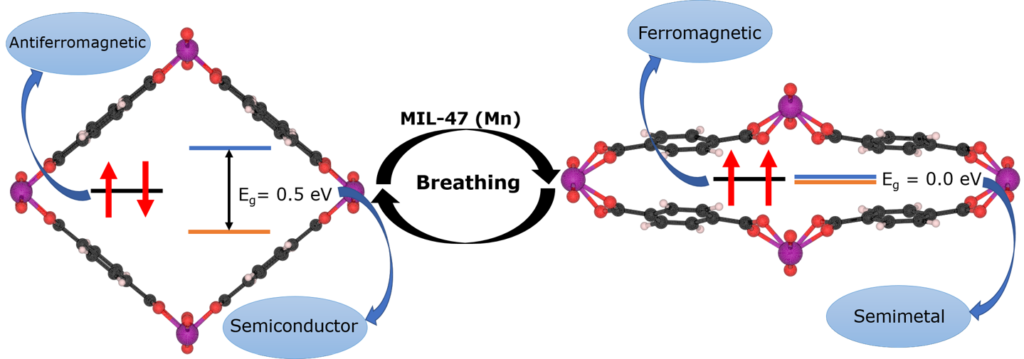
Carbon 172, 463-473 (2021),
doi: 10.1016/j.carbon.2020.10.061 {IF(2019)=8.821} -
J. Appl. Phys. 128 (5), 054901 (2020), [Featured Article][Scilight]
doi: 10.1063/5.0012285 {IF(2019)=2.286} -
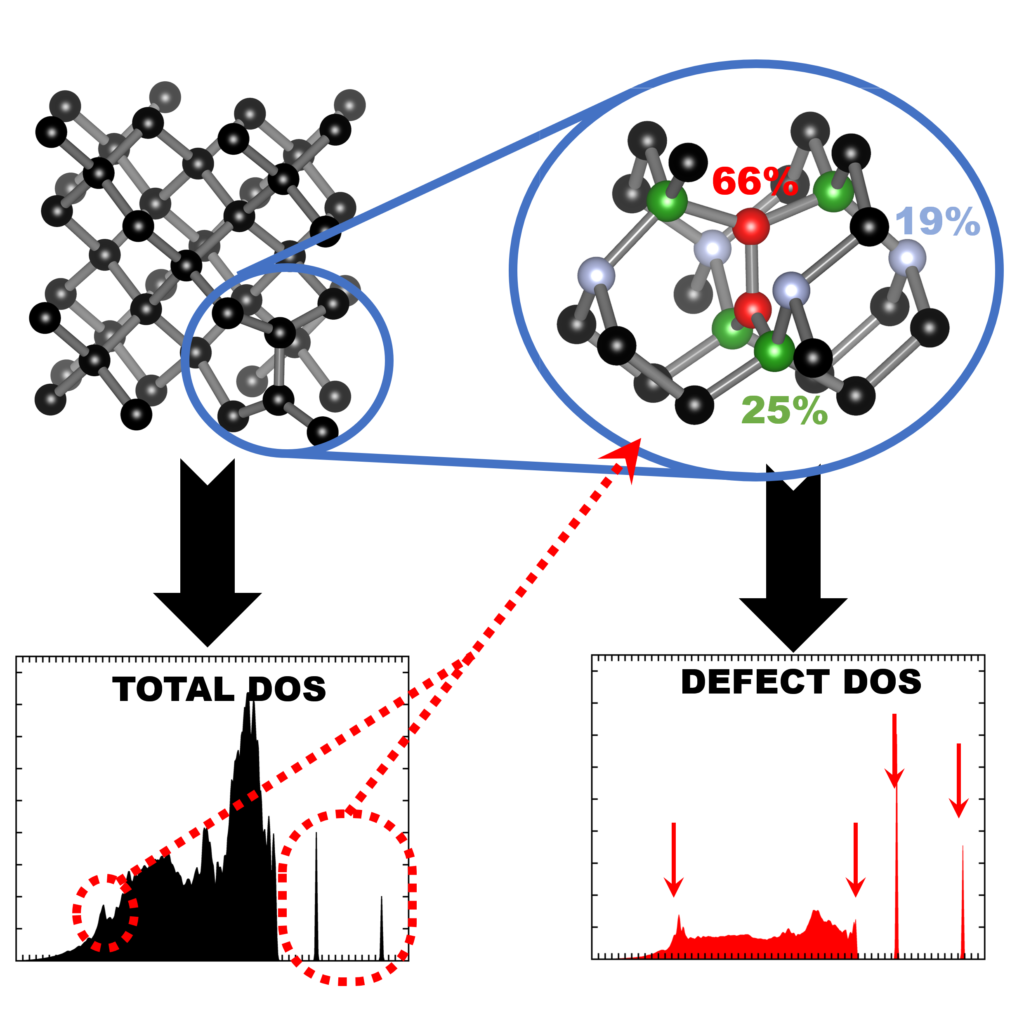
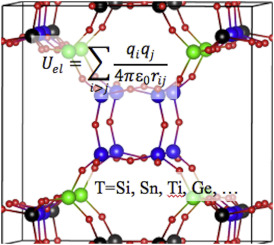
Comput. Mater. Sci. 181, 109736 (2020),
doi: 10.1016/j.commatsci.2020.109736 {IF(2019)=2.863} -
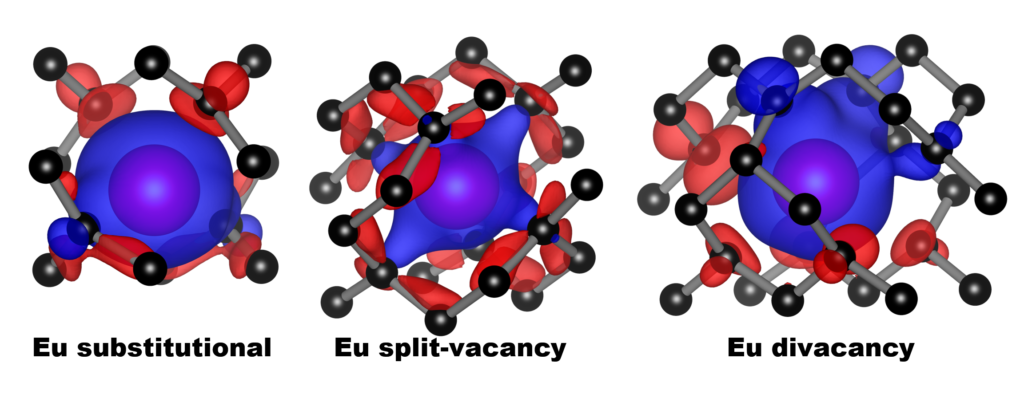
Carbon 162, 1-12 (2020),
doi: 10.1016/j.carbon.2020.01.115 {IF(2019)=8.821} -
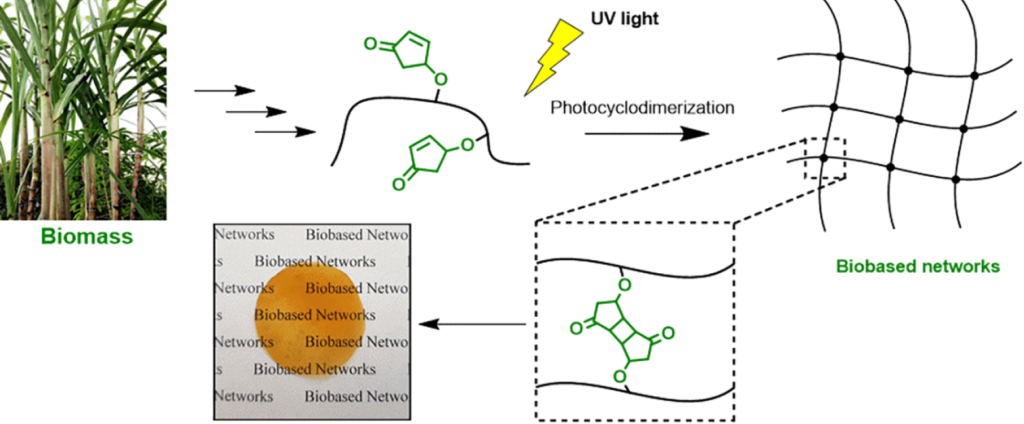
Macromolecules 53(4), 1388-1404 (2020),
doi: 10.1021/acs.macromol.9b02659 {IF(2019)=5.918} -
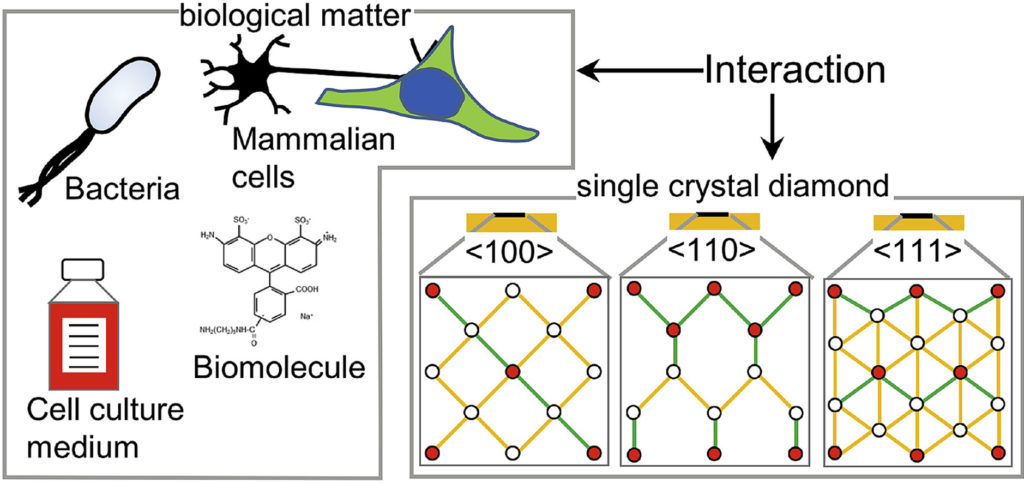
RSC Adv. 10, 4786-4794 (2020),
doi: 10.1039/C9RA09196C {IF(2019)=3.119}
2. Completed refereeing tasks: +17
- Applied Physics Letters
- Journal of Physical Chemistry (2x)
- Computational Materials Science (2x)
- Materials Chemistry and Physics
- Journal of Physics: Condensed Matter (5x)
- Diamond and Related Materials (6x)
3. Conferences & workshops in times of Corona: +3/+1 (Attended & Organised), >+4 internal

ACOS poster prize 2020
With regard to conferences, 2020 was the year everyone came into contact with the concept of the online conference. Many conferences and events got canceled: such as TEDx@UHasselt (which will return in 2021)
- ACOS 2020, Online, Oktober 28th, 2020 [poster presentation and video-pitch, 2nd poster prize]
- RSC Chemical Science Symposium 2020, Online, September 29th-30th, 2020 [iposter presentation]
- D-NL-HIT project meetings [oral presentations]
- Virtual Partner Meeting, April 8th, 2020
- Adhesives Pilot Branch meeting, October 7th, 2020
- Virtual Partner Meeting, October 15th, 2020
- UV-Curing Branch meeting, October 22nd, 2020
- SBDD XXV, Hasselt University, Belgium, March 11th-13th, 2020 [(invited) oral presentation, poster presentation] …On Friday13th Belgium went into it’s first lock-down.
- Pilot Branch meeting adhesives D-NL-HIT project, Maastricht University, Brighlands campus, February 26th, 2020 [Organised]
4. Science Communication & Social media:
- In February 2020, I finally caved and joined Twitter as @DelocalizedD .
- Added several new repositories to my github account, with the most important ones being:
- AMADEUS framework for ML of small datasets
- LS-SVM library
- Started a YouTube channel (for the ACOS video pitch)
5. Current size of HIVE:
- Continued work on a public version of HIVE at github: HIVE 4.x (26K lines, 9 commands available)
- 61K lines of program (code: 69 %)
- ~100 files
- 49 (command line) options
6. Hive-STM program:
- 32 new users in 2020 (making for a total of 407 users)