Category: publications
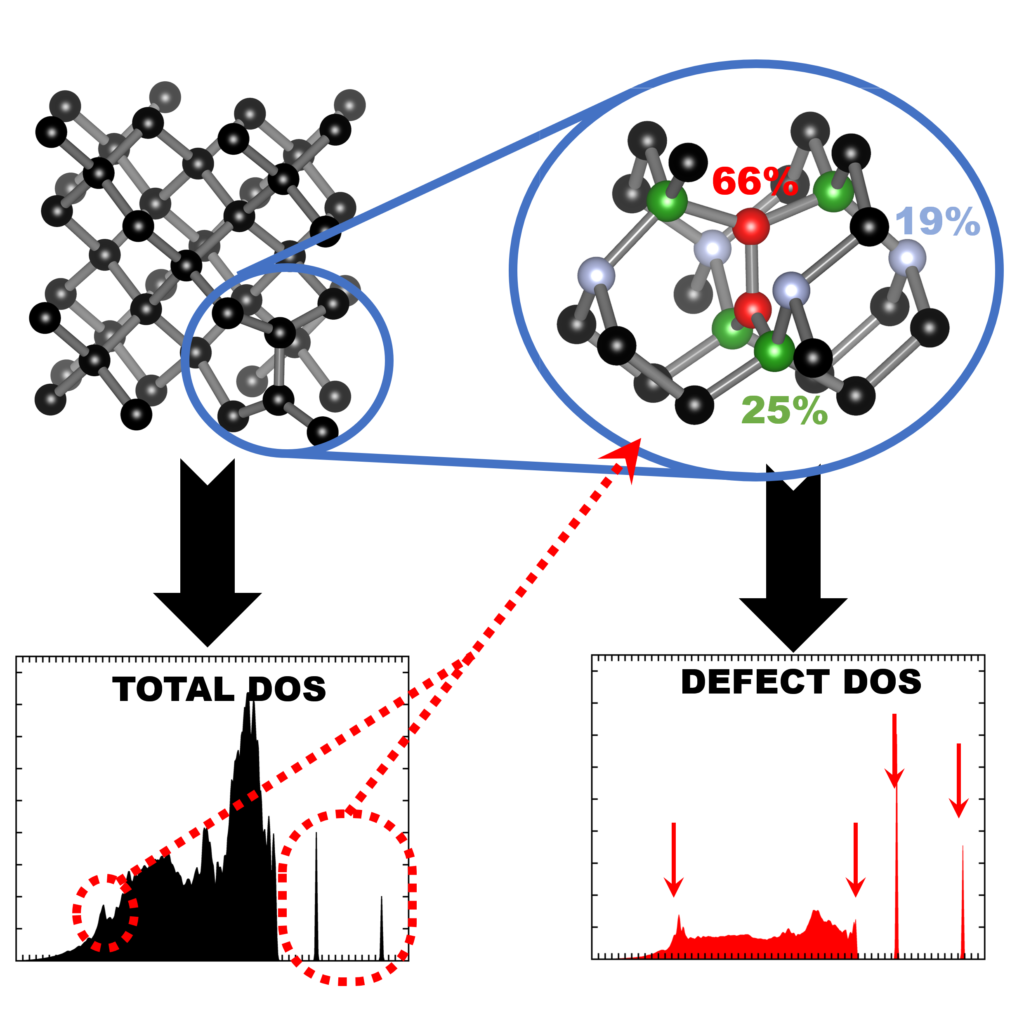 |
| Graphical Abstract: Finger printing defects in diamond through the creation of the vibrational spectrum of a defect. |
Vibrational spectroscopy techniques are some of the most-used tools for materials
characterization. Their simulation is therefore of significant interest, but commonly
performed using low cost approximate computational methods, such as force-fields.
Highly accurate quantum-mechanical methods, on the other hand are generally only used
in the context of molecules or small unit cell solids. For extended solid systems,
such as defects, the computational cost of plane wave based quantum mechanical simulations
remains prohibitive for routine calculations. In this work, we present a computational scheme
for isolating the vibrational spectrum of a defect in a solid. By quantifying the defect character
of the atom-projected vibrational spectra, the contributing atoms are identified and the strength
of their contribution determined. This method could be used to systematically improve phonon
fragment calculations. More interestingly, using the atom-projected vibrational spectra of the
defect atoms directly, it is possible to obtain a well-converged defect spectrum at lower
computational cost, which also incorporates the host-lattice interactions. Using diamond as
the host material, four point-defect test cases, each presenting a distinctly different
vibrational behaviour, are considered: a heavy substitutional dopant (Eu), two intrinsic
point-defects (neutral vacancy and split interstitial), and the negatively charged N-vacancy
center. The heavy dopant and split interstitial present localized modes at low and high
frequencies, respectively, showing little overlap with the host spectrum. In contrast, the
neutral vacancy and the N-vacancy center show a broad contribution to the upper spectral range
of the host spectrum, making them challenging to extract. Independent of the vibrational behaviour,
the main atoms contributing to the defect spectrum can be clearly identified. Recombination of
their atom-projected spectra results in the isolated spectrum of the point-defect.
Permanent link to this article: https://dannyvanpoucke.be/paper_vibrdefect-en/
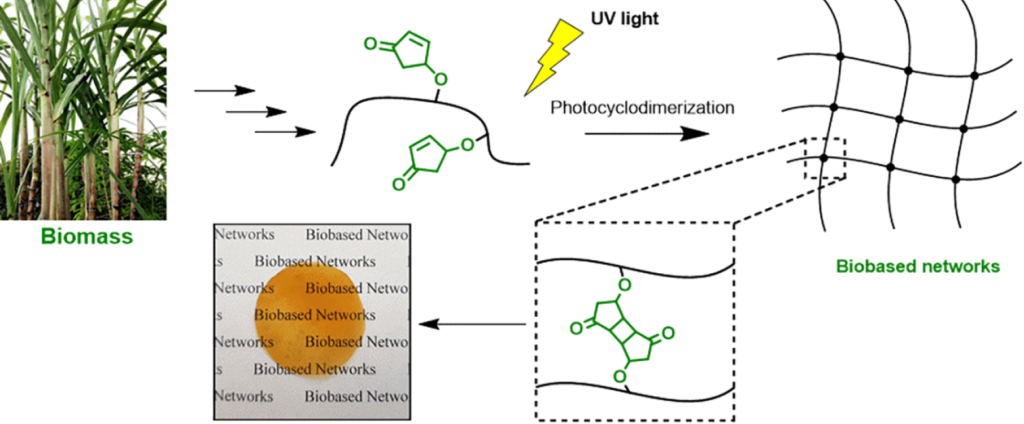 |
| Graphical Abstract: The formation of biobased polyacrylates. |
The controlled polymerization of a new biobased monomer, 4-oxocyclopent-2-en-1-yl acrylate (4CPA), was
established via reversible addition−fragmentation chain transfer (RAFT) (co)polymerization to yield polymers bearing pendent cyclopentenone units. 4CPA contains two reactive functionalities, namely, a vinyl group and an internal double bond, and is an unsymmetrical monomer. Therefore, competition between the internal double bond and the vinyl group eventually leads to gel formation. With RAFT polymerization, when aiming for a degree of polymerization (DP) of 100, maximum 4CPA conversions of the vinyl group between 19.0 and 45.2% were obtained without gel formation or extensive broadening of the dispersity. When the same conditions were applied in the copolymerization of 4CPA with lauryl acrylate (LA), methyl acrylate (MA), and isobornyl acrylate, 4CPA conversions of the vinyl group between 63 and 95% were reached. The additional functionality of 4CPA in copolymers was demonstrated by model studies with 4-oxocyclopent-2-en-1-yl acetate (1), which readily dimerized under UV light via [2 + 2] photocyclodimerization. First-principles quantum mechanical simulations supported the experimental observations made in NMR. Based on the calculated energetics and chemical shifts, a mixture of head-to-head and head-to-tail dimers of (1) were identified. Using the dimerization mechanism, solvent-cast LA and MA copolymers containing 30 mol % 4CPA were cross-linked under UV light to obtain thin films. The cross-linked films were characterized by dynamic scanning calorimetry, dynamic mechanical analysis, IR, and swelling experiments. This is the first case where 4CPA is described as a monomer for functional biobased polymers that can undergo additional UV curing via photodimerization.
Permanent link to this article: https://dannyvanpoucke.be/paper_nmrjules-en/
| Authors: |
Viraj Damle, Kaiqi Wu, Oreste De Luca, Natalia Ortí-Casañ, Neda Norouzi, Aryan Morita, Joop de Vries, Hans Kaper, Inge Zuhorn, Ulrich Eisel, Danny E.P. Vanpoucke, Petra Rudolf, and Romana Schirhagl, |
| Journal: |
Carbon 162, 1-12 (2020) |
| doi: |
10.1016/j.carbon.2020.01.115 |
| IF(2019): |
8.821 |
| export: |
bibtex |
| pdf: |
<Carbon> (Open Access) |
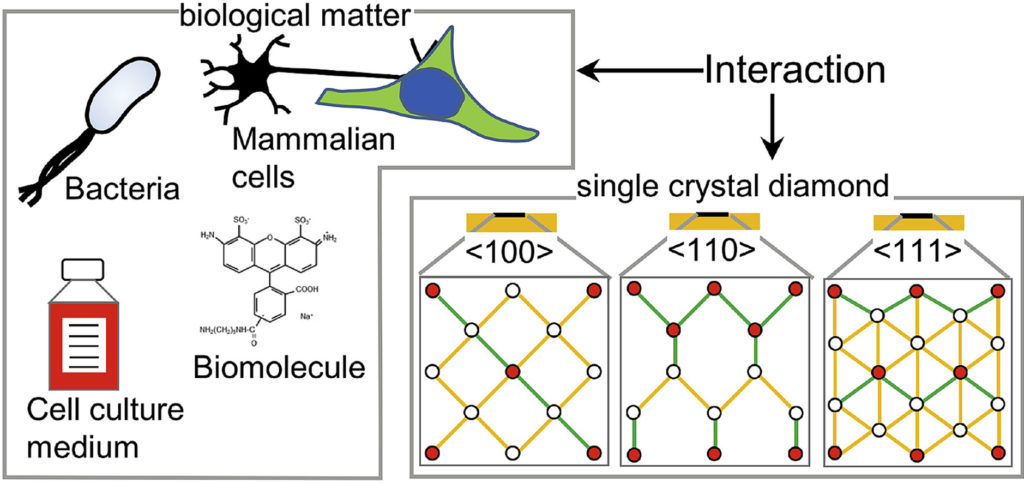 |
| Graphical Abstract: The preferential adsorption of biological matter on oriented diamond surfaces. |
Diamond has been a popular material for a variety of biological applications due to its favorable chemical, optical, mechanical and biocompatible properties. While the lattice orientation of crystalline material is known to alter the interaction between solids and biological materials, the effect of diamond’s crystal orientation on biological applications is completely unknown. Here, we experimentally evaluate the influence of the crystal orientation by investigating the interaction between the <100>, <110> and <111> surfaces of the single crystal diamond with biomolecules, cell culture medium, mammalian cells and bacteria. We show that the crystal orientation significantly alters these biological interactions. Most surprising is the two orders of magnitude difference in the number of bacteria adhering on <111> surface compared to <100> surface when both the surfaces were maintained under the same condition. We also observe differences in how small biomolecules attach to the surfaces. Neurons or HeLa cells on the other hand do not have clear preferences for either of the surfaces. To explain the observed differences, we theoretically estimated the surface charge for these three low index diamond surfaces and followed by the surface composition analysis using x-ray photoelectron spectroscopy (XPS). We conclude that the differences in negative surface charge, atomic composition and functional groups of the different surface orientations lead to significant variations in how the single crystal diamond surface interacts with the studied biological entities.
Permanent link to this article: https://dannyvanpoucke.be/paper_diamondromanaorientation2020-en/
| Authors: |
Mohammadreza Hosseini, Danny E. P. Vanpoucke, Paolo Giannozzi, Masoud Berahman and Nasser Hadipour |
| Journal: |
RSC Adv. 10, 4786-4794 (2020)
|
| doi: |
10.1039/C9RA09196C |
| IF(2019): |
3.119 |
| export: |
bibtex |
| pdf: |
<RSC Adv.> (Open Access) |
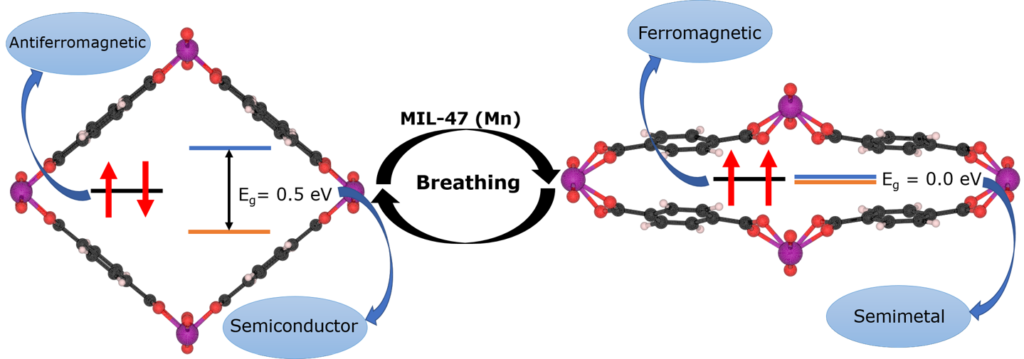 |
| Graphical Abstract: The breathing MIL-47(Mn) Metal-Organic Framework. Upon breathing, the electronic structure of this MOF undergoes a transition from an anti-ferromagnetic semiconductor, to a ferromagnetic semi-metal. |
The structural, electronic and magnetic properties of the MIL-47(Mn) metal–organic framework are investigated using first principles calculations. We find that the large-pore structure is the ground state of this material. We show that upon transition from the large-pore to the narrow-pore structure, the magnetic ground-state configuration changes from antiferromagnetic to ferromagnetic, consistent with the computed values of the intra-chain coupling constant. Furthermore, the antiferromagnetic and ferromagnetic configuration phases have intrinsically different electronic behavior: the former is semiconducting, the latter is a metal or half-metal. The change of electronic properties during breathing posits MIL-47(Mn) as a good candidate for sensing and other applications. Our calculated electronic band structure for MIL-47(Mn) presents a combination of flat dispersionless and strongly dispersive regions in the valence and conduction bands, indicative of quasi-1D electronic behavior. The spin coupling constants are obtained by mapping the total energies onto a spin Hamiltonian. The inter-chain coupling is found to be at least one order of magnitude smaller than the intra-chain coupling for both large and narrow pores. Interestingly, the intra-chain coupling changes sign and becomes five times stronger going from the large pore to the narrow pore structure. As such MIL-47(Mn) could provide unique opportunities for tunable low-dimensional magnetism in transition metal oxide systems.
Permanent link to this article: https://dannyvanpoucke.be/paper_mil47mn_moh-en/
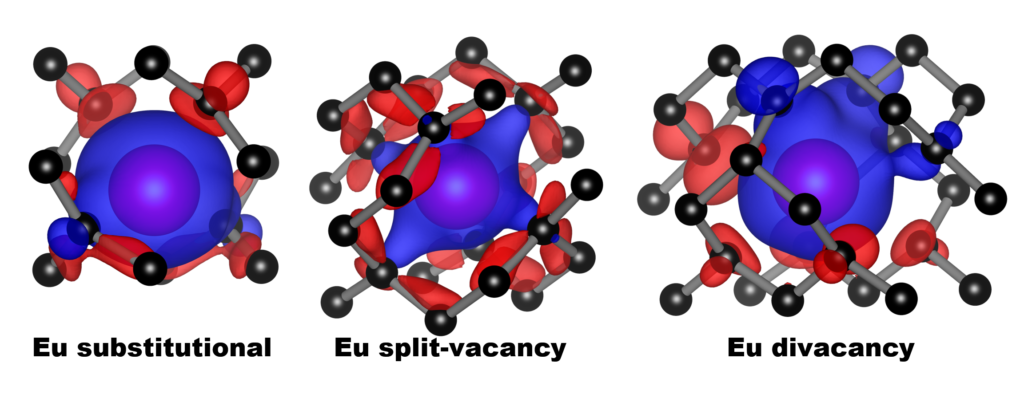 |
| Graphical Abstract: Spin polarization around the various Eu-defect models in diamond. Blue and red represent the up and down spin channels respectively. |
The incorporation of Eu into the diamond lattice is investigated in a combined theoretical-experimental study. The large size of the Eu ion induces a strain on the host lattice, which is minimal for the Eu-vacancy complex. The oxidation state of Eu is calculated to be 3+ for all defect models considered. In contrast, the total charge of the defect-complexes is shown to be negative: -1.5 to -2.3 electron. Hybrid-functional electronic-band-structures show the luminescence of the Eu defect to be strongly dependent on the local defect geometry. The 4-coordinated Eu substitutional dopant is the most promising candidate to present the typical Eu3+ luminescence, while the 6-coordinated Eu-vacancy complex is expected not to present any luminescent behaviour. Preliminary experimental results on the treatment of diamond �films with Eu-containing precursor indicate the possible incorporation of Eu into diamond �films treated by drop-casting. Changes in the PL spectrum, with the main luminescent peak shifting from approximately 614 nm to 611 nm after the growth plasma exposure, and the appearance of a shoulder peak at 625 nm indicate the potential incorporation. Drop-casting treatment with an electronegative polymer material was shown not to be necessary to observe the Eu signature following the plasma exposure, and increased the background
luminescence.
Permanent link to this article: https://dannyvanpoucke.be/paper_eudopingdrmspecial2018-en/
| Authors: |
Seyyed Amin Rounaghi, Danny E. P. Vanpoucke, Elaheh Esmaeili, Sergio Scudino, and Jürgen Eckert |
| Journal: |
J. Alloys Compd. 778, 327-336 (2019) |
| doi: |
10.1016/j.jallcom.2018.11.007 |
| IF(2019): |
4.650 |
| export: |
bibtex |
| pdf: |
<JAlloysCompd> |
Nanostructured epsilon iron carbonitride (ε-Fe3CxN1-x, x ∼ 0.05) powder with high purity (>97 wt%) was synthesized through a simple mechanochemical reaction between metallic iron and melamine. Various characterization techniques were employed to investigate the chemical and physical characteristics of the milling intermediates and the final products. The thermodynamic stability of the different phases in the Fe-C-N ternary system, including nitrogen and carbon doped structures were studied through density functional theory (DFT) calculations. A Boltzmann-distribution model was developed to qualitatively assess the stability and the proportion of the different milling products vs. milling energy. The theoretical and experimental results revealed that the milling products mainly comprise the ε-Fe3CxN1-xphase with a mean crystallite size of around 15 nm and a trace of amorphous carbonmaterial. The thermal stability and magnetic properties of the milling products were thoroughly investigated. The synthesized ε-Fe3CxN1-x exhibited thermal stabilities up to 473 K and 673 K in air and argon atmospheres, respectively, and soft magnetic properties with a saturation magnetization of around 125 emu/g.
Permanent link to this article: https://dannyvanpoucke.be/paper_epsilonfenc-en/
| Authors: |
Jarod J. Wolffis, Danny E. P. Vanpoucke, Amit Sharma, Keith V. Lawler, and Paul M. Forster |
| Journal: |
Microporous Mesoporous Mater. 277, 184-196 (2019) |
| doi: |
10.1016/j.micromeso.2018.10.028 |
| IF(2019): |
4.551 |
| export: |
bibtex |
| pdf: |
<MicroporousMesoporousMater> |
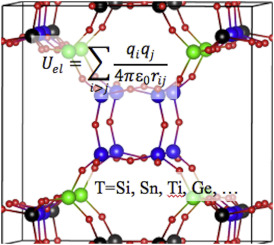 |
| Graphical Abstract: Partial charges in zeolites for force fields. |
Partial atomic charge, which determines the magnitude of the Coulombic non-bonding interaction, represents a critical parameter in molecular mechanics simulations. Partial charges may also be used as a measure of physical properties of the system, i.e. covalency, acidic/catalytic sites, etc. A range of methods, both empirical and ab initio, exist for calculating partial charges in a given solid, and several of them are compared here for siliceous (pure silica) zeolites. The relationships between structure and the predicted partial charge are examined. The predicted partial charges from different methods are also compared with related experimental observations, showing that a few of the methods offer some guidance towards identifying the T-sites most likely to undergo substitution or for proton localization in acidic framework forms. Finally, we show that assigning unique calculated charges to crystallographically unique framework atoms makes an appreciable difference in simulating predicting N2 and O2 adsorption with common dispersion-repulsion parameterizations.
Permanent link to this article: https://dannyvanpoucke.be/paper_hizeolites_2018-en/
| Authors: |
Bartłomiej M. Szyja and Danny Vanpoucke |
| Book: |
Zeolites and Metal-Organic Frameworks, (2018) |
| Chapter |
Ch 9, p 235-264 |
| Title |
Computational Chemistry Experiment Possibilities |
| ISBN: |
978-94-629-8556-8 |
| export: |
bibtex |
| pdf: |
<Amsterdam University Press>
<Open Access> |
 |
| Zeolites and Metal-Organic Frameworks (the hard-copy) |
Thanks to a rapid increase in the computational power of modern CPUs, computational methods have become a standard tool for the investigation of physico-chemical phenomena in many areas of chemistry and technology. The area of porous frameworks, such as zeolites, metal-organic frameworks (MOFs) and covalent-organic frameworks (COFs), is not different. Computer simulations make it possible, not only to verify the results of the experiments, but even to predict previously inexistent materials that will present the desired experimental properties. Furthermore, computational research of materials provides the tools necessary to obtain fundamental insight into details that are often not accessible to physical experiments.
The methodology used in these simulations is quite specific because of the special character of the materials themselves. However, within the field of porous frameworks, density functional theory (DFT) and force fields (FF)
are the main actors. These methods form the basis of most computational studies, since they allow the evaluation of the potential energy surface (PES) of the system.
Newsflash: here
Permanent link to this article: https://dannyvanpoucke.be/chaptermofs_2018-en/
One of the new features provided by Elsevier upon publication is the creation of audioslides. This is a kind of short presentation of the publication by one of the authors. I have been itching to try this since our publication on the neutral C-vancancy was published. The interface is quite intuitive, although the adobe flash tend to have a hard time finding the microphone. However, once it succeeds, things go quite smoothly. The resolution of the slides is a bit low, which is unfortunate (but this is only for the small-scale version, the large-scale version is quite nice as you can see in the link below). Maybe I’ll make a high resolution version video and put it on Youtube, later.
The result is available here (since the embedding doesn’t play nicely with WP).
And a video version can be found here.
Permanent link to this article: https://dannyvanpoucke.be/audioslides-tryout-en/
| Authors: |
Danny E. P. Vanpoucke and Ken Haenen |
| Journal: |
Diam. Relat. Mater 79, 60-69 (2017) |
| doi: |
10.1016/j.diamond.2017.08.009 |
| IF(2017): |
2.232 |
| export: |
bibtex |
| pdf: |
<DiamRelatMater> |
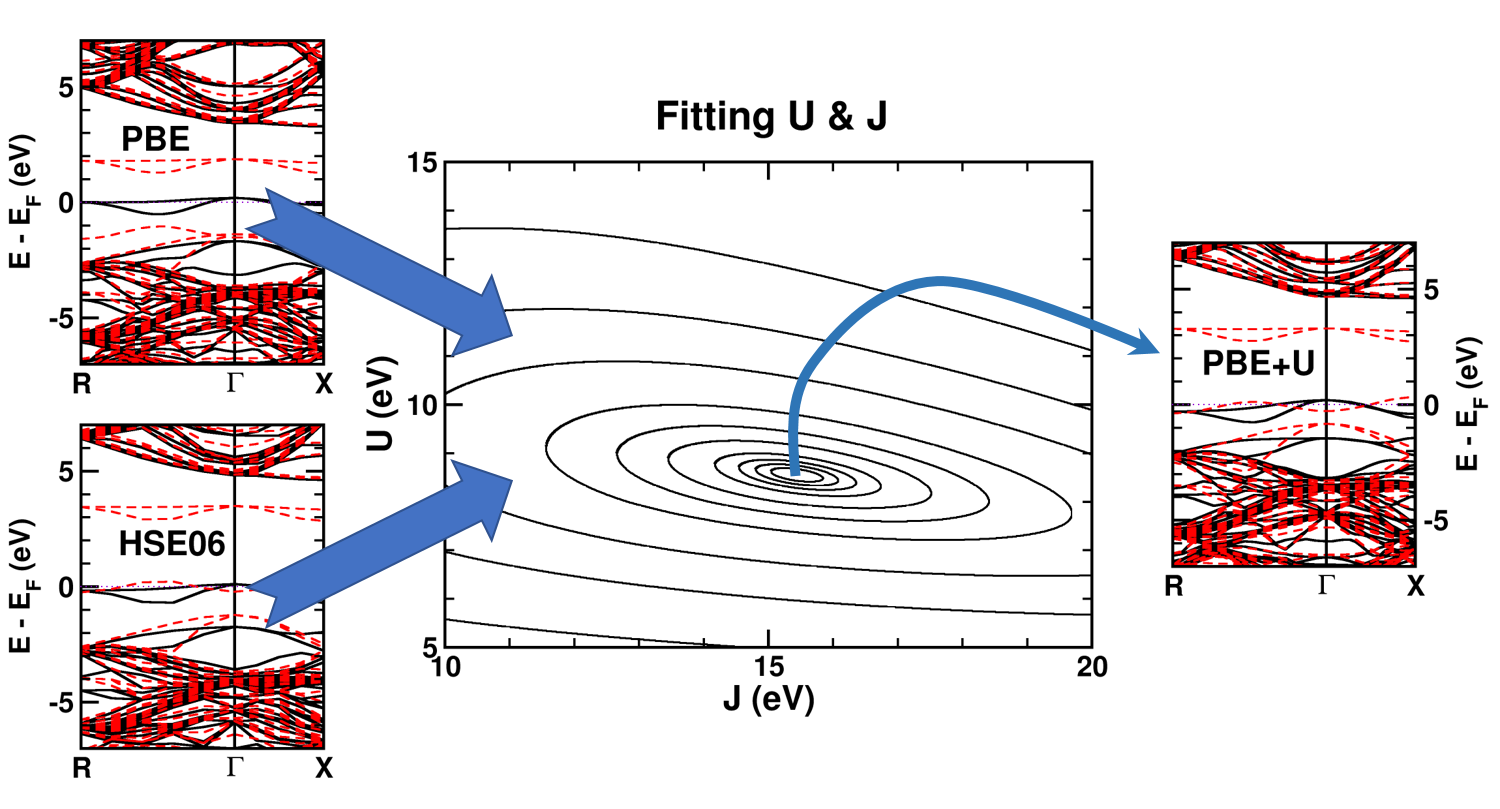 |
| Graphical Abstract: Combining a scan over possible values for U and J with reference electronic structures obtained using the hybrid functional HSE06, DFT+U can be fit to provide hybrid functional quality electronic structures at the cost of DFT calculations. |
The neutral C-vacancy is investigated using density functional theory calculations. We show that local functionals, such as PBE, can predict the correct stability order of the different spin states, and that the success of this prediction is related to the accurate description of the local magnetic configuration. Despite the correct prediction of the stability order, the PBE functional still fails predicting the defect states correctly. Introduction of a fraction of exact exchange, as is done in hybrid functionals such as HSE06, remedies this failure, but at a steep computational cost. Since the defect states are strongly localized, the introduction of additional on site Coulomb and exchange interactions, through the DFT+U method, is shown to resolve the failure as well, but at a much lower computational cost. In this work, we present optimized U and J parameters for DFT+U calculations, allowing for the accurate prediction of defect states in defective
diamond. Using the PBE optimized atomic structure and the HSE06 optimized electronic structure as reference, a pair of on-site Coulomb and exchange parameters (U,J) are fitted for DFT+U studies of defects in diamond.
Poster-presentation: here
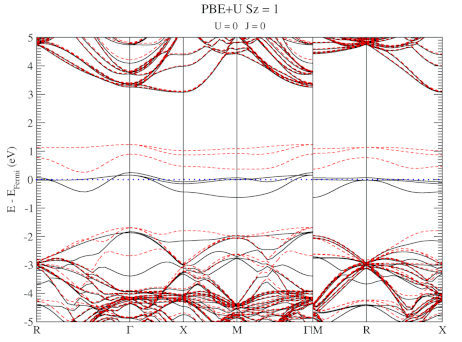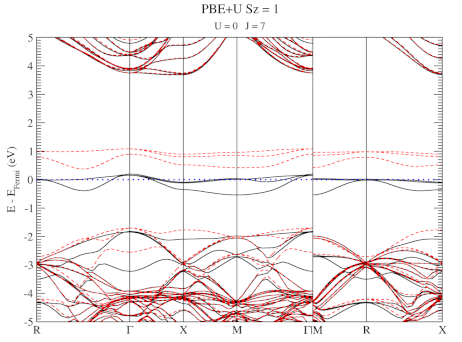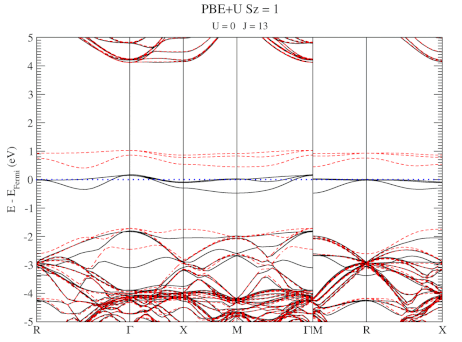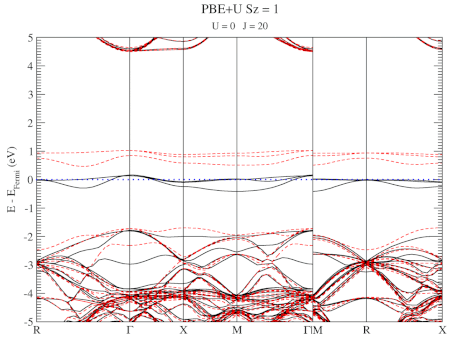
DFT+U series (varying J) for a specific spin state of the C-vacancy defect.
Permanent link to this article: https://dannyvanpoucke.be/paperdrm-diamond-dftu-en/





