 “I have a question”(ik heb een vraag). This is the name of a Belgian (Flemisch) website aimed at bringing Flemisch scientists and the general public together through scientific or science related questions. The basic idea is rather simple. Someone has a scientific question and poses it on this website, and a scientist will provide an answer. It is an excellent opportunity for the latter to hone his/her own science communication skills (and do some outreach) and for the former to get an good answer to his/her question.
“I have a question”(ik heb een vraag). This is the name of a Belgian (Flemisch) website aimed at bringing Flemisch scientists and the general public together through scientific or science related questions. The basic idea is rather simple. Someone has a scientific question and poses it on this website, and a scientist will provide an answer. It is an excellent opportunity for the latter to hone his/her own science communication skills (and do some outreach) and for the former to get an good answer to his/her question.
All questions and answers are collected in a searchable database, which currently contains about fifteen thousand questions answered by a (growing) group of nearly one thousand scientists. This is rather impressive for a region of about 6.5 Million people. I recently joined the group of scientists providing answers.
An interesting materials-related question was posed by Denis (my translation of his question and context):
What is the relation between the density of a material and its thermal expansion?
I was wondering if there exists a relation between the density of a material and the thermal expansion (at the same temperature)? In general, gasses expand more than solids, so can I extend this to the following: Materials with a small density will expand more because the particles are separated more and thus experience a small cohesive force. If this statement is true, then this would imply that a volume of alcohol should expand more than the same volume of air, which I think is puzzling. Can you explain this to me?
Answer (a bit more expanded than the Dutch one):
Unfortunately there exists no simple relation between the density of a material and its thermal expansion coefficient.
Let us first correct something in the example given: the density of alcohol (or ethanol) is 46.07 g/mol (methanol would be 32.04 g/mol) which is significantly more than the density of air which is 28.96 g/mol. So following the suggested assumption, air should expand more. If we look at liquids, it is better to compare ethanol (0.789 g/cm3) to compare water (1 g/cm3) as liquid air (0.87 g/cm3) needs to be cooled below -196 °C (77K). The thermal expansion coefficients of wtare and ethanol are 207×10-6/°C and 750×10-6/°C, respectively. So in this case, we see that alcohol will expand more than water (at 20°C). Supporting Denis’ statement.
Unfortunately, these are just two simple materials at a very specific temperature for which this statement is true. In reality, there are many interesting aspects complicating life. A few things to keep in mind are:
- A gas (in contrast to a liquid or solid) has no own boundary. So if you do not put it in any type of a container, then it will just keep expanding. The change in volume observed when a gas is heated is due to an increase in pressure (the higher kinetic energy of the gas molecules makes them bounce harder of the walls of your container, which can make a piston move or a balloon grow). In a liquid or a solid on the other hand, the expansion is rather a stretching of the material itself.
- Furthermore, the density does not play a role at all, in case of the expansion of an ideal gas, since p*V=n*R*T. From this it follows that 1 mole of H2 gas, at 20°C and a pressure of 1 atmosphere, has the exact same volume as 1 mole of O2 gas, at 20°C and a pressure of 1 atmosphere, even though the latter has a density which is 16 times higher.
- There are quite a lot of materials which show a negative thermal expansion in a certain temperature region (i.e. they shrink when you increase the temperature). One well-known example is water. The density of liquid water at 0 °C is lower than that of water at 4 °C. This is the reason why there remains some liquid water at the bottom of a pond when it is frozen over.
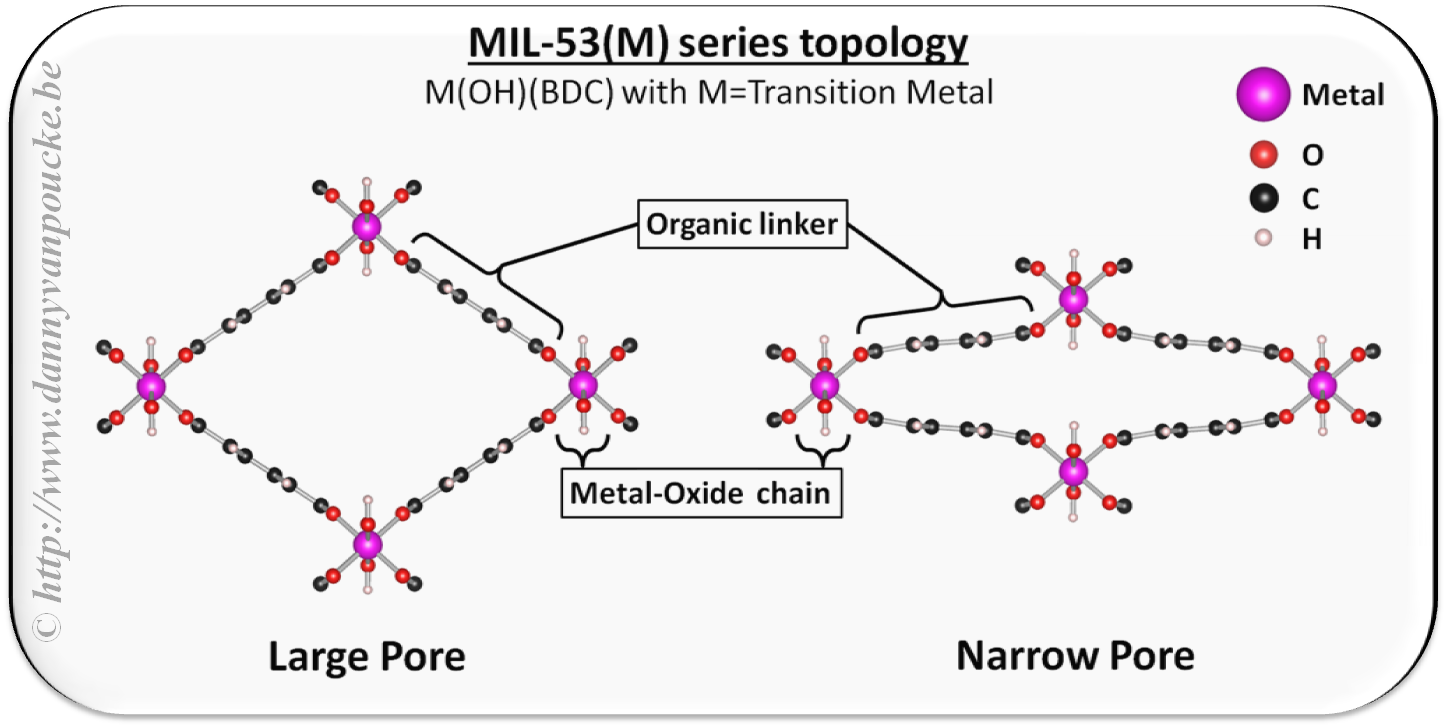
- There are also materials which show “breathing” behavior (this are reversible volume changes in solids which made the originators of the term think of human breathing: inhaling expands our lungs and chest, while exhaling contracts it again.) One specific class of these materials are breathing Metal-Organic Frameworks (MOFs). Some of these look like wine-racks (see figure here) which can open and close due to temperature variations. These volume variations can be 50% or more! 😯
The way a material expands due to temperature variations is a rather complex combination of different aspects. It depends on how thermal vibrations (or phonons) propagate through the material, but also on the possible presence of phase-transitions. In some materials there are even phase-transitions between solid phases with a different crystal structure. These, just like solid/liquid phase transitions can lead to very sudden jumps in volume during heating or cooling. These different crystal phases can also have very different physical properties. During the middle-ages, tin pest was a large source of worries for organ-builders. At a temperature below 13°C β-tin is more stable α-tin, which is what was used in organ pipes. However, the high activation energy prevents the phase-transformation from α-tin to β-tin to happen too readily. At temperatures of -30 °C and lower this barrier is more easily overcome.This phase-transition gives rise to a volume reduction of 27%. In addition, β-tin is also a brittle material, which easily disintegrates. During the middle ages this lead to the rapid deterioration and collapse of organ-pipes in church organs during strong winters. It is also said to have caused the buttons of the clothing of Napoleon’s troops to disintegrate during his Russian campaign. As a result, the troops’ clothing fell apart during the cold Russian winter, letting many of them freeze to death.





 Today was the first day of the
Today was the first day of the  Ball-and-stick model of zinc gallate (right) density of states of Cr doped zinc gallate.")
{kind=link}