Today VLIR (Flemish Inter-university Council) and the Young Academy had a conference on the future of fundamental research in Flanders: Roots of Science. We live in a world where we rely on science more and more to resolve our problems (think climate change, disease control, energy generation, …). In our bizarre world of alternative facts and fake news, science can be utterly ignored in one sentence and proposed as a magical solution in the next.
Although I am happy with the faith some have in the possibilities of science, it is important to remember that it is not magic. This has a very important consequence:
Things do not happen simply because you want them to happen.
Many important breakthroughs in science are what one would call serendipity (e.g., the discovery of penicillin by Fleming, development of the WWW as a side-effect of researchers wanting to share their data

at CERN in 1991,…) . In Flanders the Royal Flemish Academy and the Young Academy have written a Standpoint (an evidence-based advisory text)
discussing the need for more researcher-driven research in contrast to agenda-driven research, as they believe this is a conditio sine qua non for a healthy scientific future.
Where government-driven research focuses on resolving questions from society, researcher-driven research allows the researcher to follow his or her personal interest. This not with the primal aim of having short-te
rm return of investment, but with the aim of providing the fundamental knowledge and expertise which some day may be needed for the former. In researcher-driver research, the journey is the goal as this is where scientific progress is made by finding solutions for problems not imagined before.
Do we have to pay for this with our tax-payers money? I think we do. No-one imagined optical drives (CD, DVD, blue-ray) to become a billion euro industry while the laser was being developed in a lab. Who would have thought the transistor would play such an important role in our every-day life? And what about the first computer? Thomas Watson, President of IBM, has allegedly said in 1943: “I think there is a world market for maybe 5 computers.” And, yet, now many of us have more than 5 computers at home (including tables, smartphones,…)! The researchers working on these “inventions” did not do this with your Blue-ray player or smartphone in mind. These high impact applications are “merely” side-products of their fundamental scientific research. No-one at the time could predict this, so why should we be able to do this today? In this sense, you should see funding of fundamental research as a long term investment. Tax-money is being invested in our future, and the future of children and grandchildren. Although we do not know what will be the outcome, we know from the past that it will have an impact on our lives.
Its difficult to make predictions, especially about the future.
Let us therefor support more researcher-driven research.
In addition to the Standpoint, there is also a very nice video explaining the situation (with subtitles in English or Dutch, use the cogwheel to select your preference).
 This summer, I had the pleasure of being interviewed by
This summer, I had the pleasure of being interviewed by 
 The power of models in physics, originates from keeping only the most important and relevant aspects. Such approximations provide a simplified picture and allow us to understand the driving forces behind nature itself. However, in this context,
The power of models in physics, originates from keeping only the most important and relevant aspects. Such approximations provide a simplified picture and allow us to understand the driving forces behind nature itself. However, in this context, 

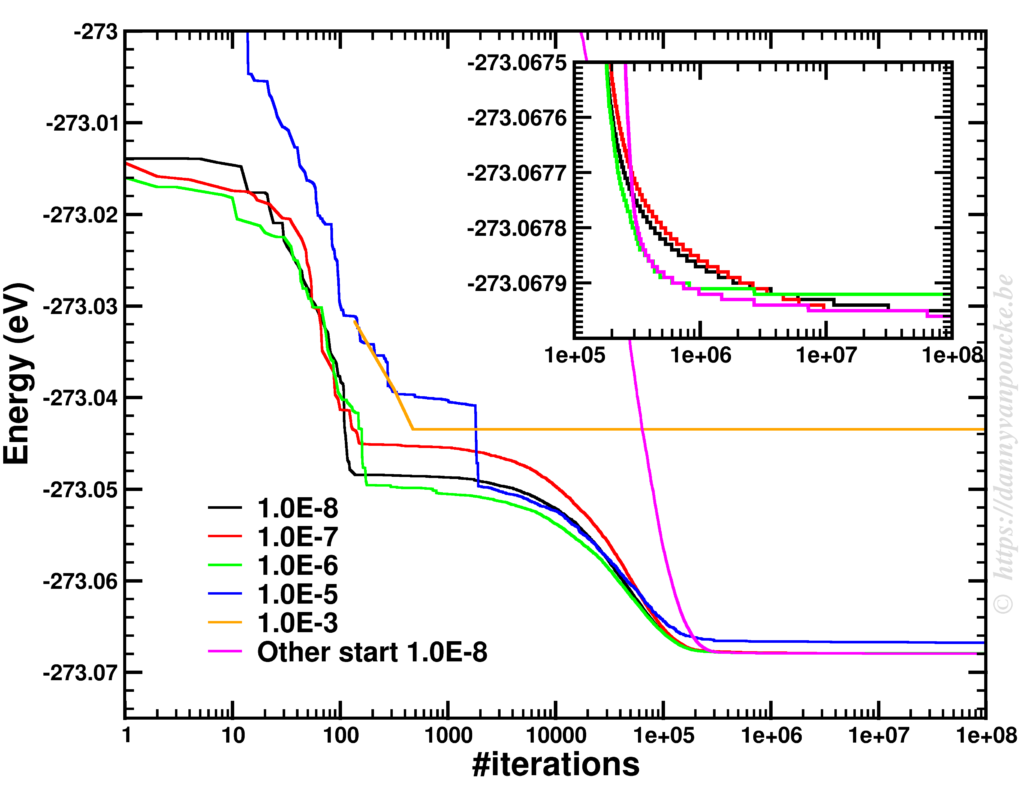
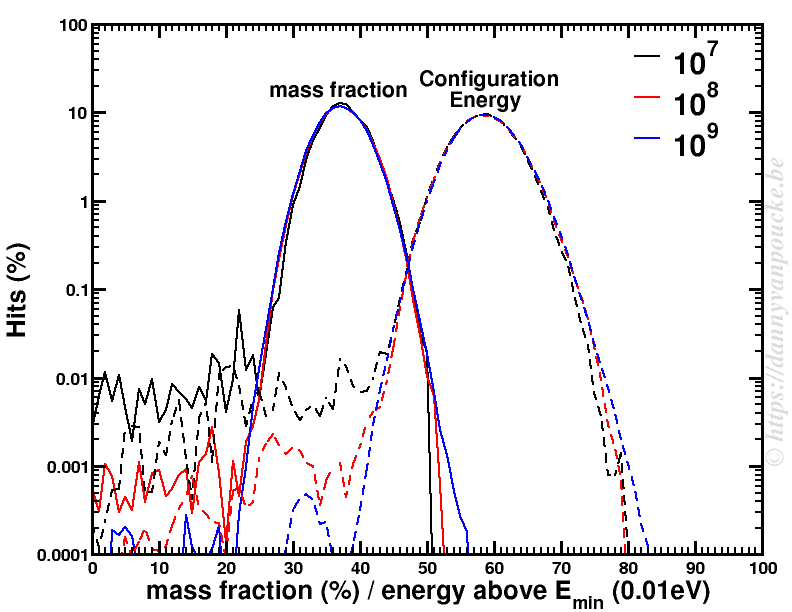


 About 10 years ago, at the end of 2007 and beginning of 2008, the 5 Flemish universities founded the Flemish Supercomputer Center (
About 10 years ago, at the end of 2007 and beginning of 2008, the 5 Flemish universities founded the Flemish Supercomputer Center (