Category: publications
 |
| Graphical Abstract: Schematic representation of the electrostatic potential within a water molecule along the lines between the atoms. The color background shows the electrostatic potential on the 0.001 a.u. contour of the density. The Vs,min and Vs,max points on the surface are indicated. |
This paper discusses the use of the electrostatic potential in both recent and older literature, with an emphasis upon a 2022 Molecular Physics article by Politzer and Murray entitled “Atoms do exist in molecules: analysis using electrostatic potentials at nuclei“. We discuss electrostatic potentials at nuclei and how they easily lead to atoms in molecules, without physically separating the individual atoms. We further summarize the work by the Politzer group on definitions of atomic radii by means of the electrostatic potential. The earlier studies began in the 1970’s and continued through the 1990’s. Unfortunately, access to these older publications is often limited, cfr. digital libraries often limit the authorized access until a certain publication year, and these papers are often not cited in current publications. Although still being highly interesting and relevant, this older literature is in danger of being lost. Digging into this older literature thus opens up new views. Our feeling is that Peter passed ‘on’ a vision that boundaries do not exist between atoms in molecules, but that some useful and meaningful radii can be obtained using the electrostatic potential between atoms in molecules.
Permanent link to this article: https://dannyvanpoucke.be/2025-paper-esp-politzer-rev-en/
| Authors: |
Thijs G.I. van Wijk, E. Aylin Melan, Rani Mary Joy, Emerick Y. Guillaume, Paulius Pobedinskas, Ken Haenen, and Danny E.P. Vanpoucke |
| Journal: |
Carbon 234, 119928 (2025) |
| doi: |
10.1016/j.carbon.2024.119928 |
| IF(2024): |
10.5 |
| export: |
bibtex |
| pdf: |
<Carbon> |
 |
| Graphical Abstract: Schematic representation of the impact of hydrostatic and linear strain on the Zero Phonon Line of the neutral GeV defect in diamond. |
Color centers in diamond, such as the GeV center, are promising candidates for quantum-based applications. Here, we investigate the impact of strain on the zero-phonon line (ZPL) position of GeV0. Both hydrostatic and linear strain are modeled using density functional theory for GeV0concentrations of 1.61 % down to 0.10 %. We present qualitative and quantitative differences between the two strain types: for hydrostatic tensile and compressive strain, red- and blue-shifted ZPL positions are expected, respectively, with a linear relation between the ZPL shift and the experienced stress. By calculating the ZPL shift for varying GeV0 concentrations, a shift of 0.15 nm/GPa (0.38 meV/GPa) is obtained at experimentally relevant concentrations using a hybrid functional. In contrast, only red-shifted ZPL are found for tensile and compressive linear strain along the ⟨100⟩ direction. The calculated ZPL shift exceeds that of hydrostatic strain by at least one order of magnitude, with a significant difference between tensile and compressive strains: 3.2 and 4.8 nm/GPa (8.1 and 11.7 meV/GPa), respectively. In addition, a peak broadening is expected
due to the lifted degeneracy of the GeV0 eg state, calculated to be about 6 meV/GPa. These calculated results are placed in perspective with experimental observations, showing values of ZPL shifts and splittings of comparable magnitude.
Permanent link to this article: https://dannyvanpoucke.be/2025-paper-strainedgev-en/
| Authors: |
Asif Iqbal Bhatti, Sandeep Kumar, Catharina Jaeken, Michael Sluydts, Danny E.P. Vanpoucke, and Stefaan Cottenier |
| Journal: |
Journal of Materials Chemistry A 13, 526-539 (2025) |
| doi: |
10.1039/D4TA06603K |
| IF(2024): |
10.7 |
| export: |
bibtex |
| pdf: |
<J.Mat.Chem.A> |
 |
| Graphical Abstract: Schematic representation of the LPS material and the variation of results obtained due to slight changes in settings within a High Throughput workflow. |
High-throughput computational screening has become a powerful tool in materials science for identifying promising candidates for specific applications. However, the effectiveness of these methods relies heavily on the accuracy and appropriateness of the underlying models and assumptions. In this study, we use the popular argyrodite solid-state electrolyte family Li6PS5X (X = Cl, Br, I) as a case study to critically examine key steps in high-throughput workflows and highlight potential pitfalls. We demonstrate some of these pitfalls by highlighting the importance of careful structural considerations, including symmetry breaking and site disorder, and examine the difference between 0 K thermodynamic stability and finite-temperature stability based on temperature-dependent Gibbs free energy calculations. Furthermore, we explore the implications of these findings for the ranking of candidate materials in a mini-throughput study in a search space of isovalent analogs to Li6PS5Cl. As a result of these findings, our work underscores the need for balanced trade-offs between computational efficiency and accuracy in high-throughput screenings, and offers guidance for designing more robust workflows that can better bridge the gap between computational predictions and experimental realities.
Permanent link to this article: https://dannyvanpoucke.be/2025-paper-thedevilinthedetails-en/
| Authors: |
Emerick Y. Guillaume, Danny E. P. Vanpoucke, Rozita Rouzbahani, Luna Pratali Maffei, Matteo Pelucchi, Yoann Olivier, Luc Henrard, & Ken Haenen |
| Journal: |
Carbon 222, 118949 (2024) |
| doi: |
10.1016/j.carbon.2024.118949 |
| IF(2022): |
10.9 |
| export: |
bibtex |
| pdf: |
<Carbon> |
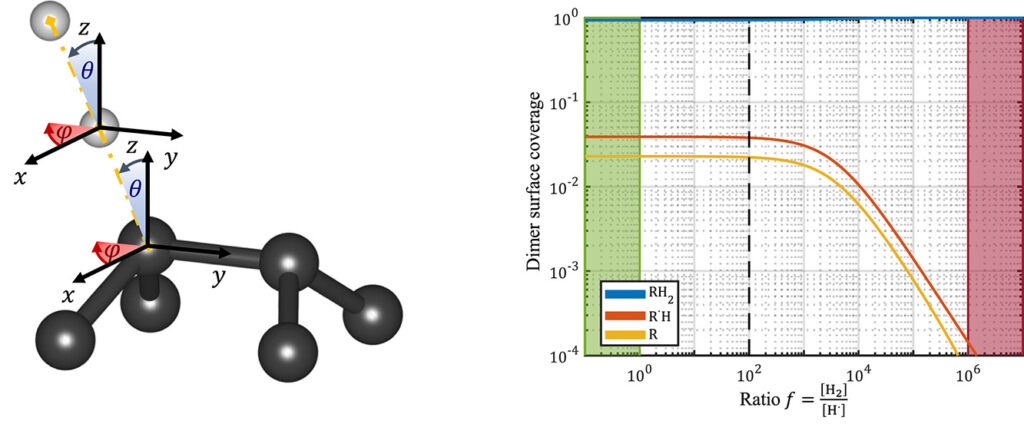 |
| Graphical Abstract: (left) Ball-and-stick representation of aH adsorption/desorption reaction mediated through a H radical. (right) Monte Carlo estimates of the H coverage of the diamond surface at different temperatures based on quantum mechanically determined reaction barriers and reaction rates. |
Hydrogen radical attacks and subsequent hydrogen migrations are considered to play an important role in the atomic-scale mechanisms of diamond chemical vapour deposition growth. We perform a comprehensive analysis of the reactions involving H-radical and vacancies on H-passivated diamond surfaces exposed to hydrogen radical-rich atmosphere. By means of first principles calculations—density functional theory and climbing image nudged elastic band method—transition states related to these mechanisms are identified and characterised. In addition, accurate reaction rates are computed using variational transition state theory. Together, these methods provide—for a broad range of temperatures and hydrogen radical concentrations—a picture of the relative likelihood of the migration or radical attack processes, along with a statistical description of the hydrogen coverage fraction of the (100) H-passivated surface, refining earlier results via a more thorough analysis of the processes at stake. Additionally, the migration of H-vacancy is shown to be anisotropic, and occurring preferentially across the dimer rows of the reconstructed surface. The approach used in this work can be generalised to other crystallographic orientations of diamond surfaces or other semiconductors.
Permanent link to this article: https://dannyvanpoucke.be/2024-paper-hadsorption-emerick-en/
| Authors: |
Emanuele Bosoni, Louis Beal, Marnik Bercx, Peter Blaha, Stefan Blügel, Jens Bröder, Martin Callsen, Stefaan Cottenier, Augustin Degomme, Vladimir Dikan, Kristjan Eimre, Espen Flage-Larsen, Marco Fornari, Alberto Garcia, Luigi Genovese, Matteo Giantomassi, Sebastiaan P. Huber, Henning Janssen, Georg Kastlunger, Matthias Krack, Georg Kresse, Thomas D. Kühne, Kurt Lejaeghere, Georg K. H. Madsen, Martijn Marsman, Nicola Marzari, Gregor Michalicek, Hossein Mirhosseini, Tiziano M. A. Müller, Guido Petretto, Chris J. Pickard, Samuel Poncé, Gian-Marco Rignanese, Oleg Rubel, Thomas Ruh, Michael Sluydts, Danny E.P. Vanpoucke, Sudarshan Vijay, Michael Wolloch, Daniel Wortmann, Aliaksandr V. Yakutovich, Jusong Yu, Austin Zadoks, Bonan Zhu, and Giovanni Pizzi |
| Journal: |
Nature Reviews Physics 6(1), (2024) |
| doi: |
web only |
| IF(2021): |
36.273 |
| export: |
NA |
| pdf: |
<NatRevPhys> |
The cover of this issue shows an artistic representation of the equations of state of the periodic table elements, calculated using two all-electron codes in each of the 10 crystal structure configurations shown on the table. The cover image is based on the Perspective Article How to verify the precision of density-functional-theory implementations via reproducible and universal workflows by E. Bosoni et al., https://doi.org/10.1038/s42254-023-00655-3. (The related paper can be found here.)

Cover Nature Reviews Physics: Accuracy of DFT modeling in solids
Permanent link to this article: https://dannyvanpoucke.be/paper2024_accuracycover-en/
| Authors: |
Emanuele Bosoni, Louis Beal, Marnik Bercx, Peter Blaha, Stefan Blügel, Jens Bröder, Martin Callsen, Stefaan Cottenier, Augustin Degomme, Vladimir Dikan, Kristjan Eimre, Espen Flage-Larsen, Marco Fornari, Alberto Garcia, Luigi Genovese, Matteo Giantomassi, Sebastiaan P. Huber, Henning Janssen, Georg Kastlunger, Matthias Krack, Georg Kresse, Thomas D. Kühne, Kurt Lejaeghere, Georg K. H. Madsen, Martijn Marsman, Nicola Marzari, Gregor Michalicek, Hossein Mirhosseini, Tiziano M. A. Müller, Guido Petretto, Chris J. Pickard, Samuel Poncé, Gian-Marco Rignanese, Oleg Rubel, Thomas Ruh, Michael Sluydts, Danny E.P. Vanpoucke, Sudarshan Vijay, Michael Wolloch, Daniel Wortmann, Aliaksandr V. Yakutovich, Jusong Yu, Austin Zadoks, Bonan Zhu, and Giovanni Pizzi |
| Journal: |
Nature Reviews Physics 6(1), 45-58 (2024) |
| doi: |
10.1038/s42254-023-00655-3 |
| IF(2021): |
36.273 |
| export: |
bibtex |
| pdf: |
<NatRevPhys>
<ArXiv:2305.17274> |
 |
| Graphical Abstract: “We hope our dataset will be a reference for the field for years to come,” says Giovanni Pizzi, leader of the Materials Software and Data Group at the Paul Scherrer Institute PSI, who led the study. (Image: Paul Scherrer Insitute / Giovanni Pizzi) |
Density-functional theory methods and codes adopting periodic boundary conditions are extensively used in condensed matter physics and materials science research. In 2016, their precision (how well properties computed with different codes agree among each other) was systematically assessed on elemental crystals: a first crucial step to evaluate the reliability of such computations. In this Expert Recommendation, we discuss recommendations for verification studies aiming at further testing precision and transferability of density-functional-theory computational approaches and codes. We illustrate such recommendations using a greatly expanded protocol covering the whole periodic table from Z = 1 to 96 and characterizing 10 prototypical cubic compounds for each element: four unaries and six oxides, spanning a wide range of coordination numbers and oxidation states. The primary outcome is a reference dataset of 960 equations of state cross-checked between two all-electron codes, then used to verify and improve nine pseudopotential-based approaches. Finally, we discuss the extent to which the current results for total energies can be reused for different goals.
Permanent link to this article: https://dannyvanpoucke.be/paper-aiidaconsortium2023-en/
| Authors: |
Ahmed M. Rozza, Danny E. P. Vanpoucke, Eva-Maria Krammer, Julie Bouckaert, Ralf Blossey, Marc F. Lensink, Mary Jo Ondrechen, Imre Bakó, Julianna Oláh, and Goedele Roos |
| Journal: |
Journal of Molecular Liquids 384, 122172 (2023) |
| doi: |
10.1016/j.molliq.2023.122172 |
| IF(2021): |
6.633 |
| export: |
bibtex |
| pdf: |
<JMolLiq> |
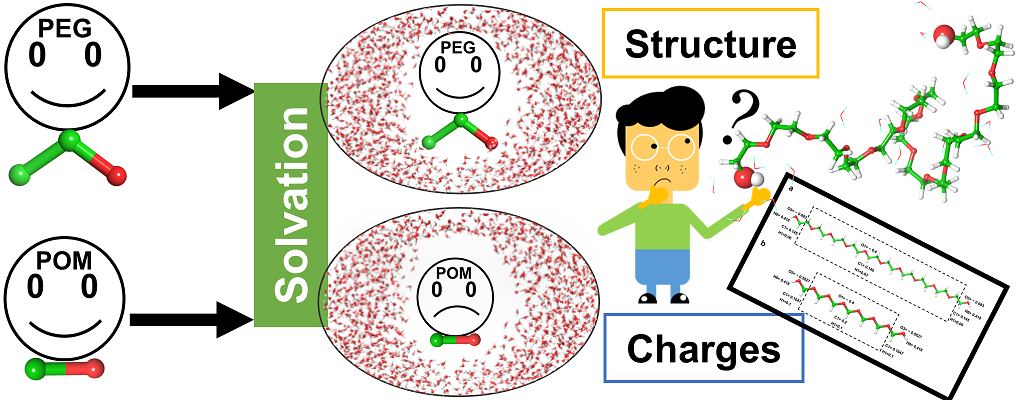 |
| Graphical Abstract: PEG or POM, similar in structure though very different in their solvation. Is this due to structure or charge(distribution)? |
Non-toxic, chemically inert, organic polymers as polyethylene glycol (PEG) and polyoxymethylene (POM) have versatile applications in basic research, industry and pharmacy. In this work, we aim to characterize the hydration structure of PEG and POM oligomers by exploring how the solute disturbs the water structure compared to the bulk solvent and how the solute chain interacts with the solvent. We explore the effect of (i) the C-C-O (PEG) versus CO (POM) constitution of the chain and (ii) chain length. To this end, MD simulations followed by clustering and topological analysis of the hydration network, as well as by quantum
mechanical calculations of atomic charges are used. We show that the hydration varies with chain conformation and length. The degree of folding of the chain impacts its degree of solvation, which is measurable by different parameters as for example the number of water molecules in the first solvation shell and the solvent accessible surface. Atomic charges calculated on the oligomers in gas phase are stable throughout conformation and chain length and seem not to determine solvation. Hydration however induces charge transfer from the solute molecule to the solvent, which depends on the degree of hydration.
Permanent link to this article: https://dannyvanpoucke.be/paper-roosg_peg-en/
| Authors: |
Danny E.P. Vanpoucke, Marie A.F. Delgove, Jules Stouten, Jurrie Noordijk, Nils De Vos, Kamiel Matthysen, Geert G.P. Deroover, Siamak Mehrkanoon, and Katrien V. Bernaerts |
| Journal: |
Polymer International 71(8), i-i (2022) |
| doi: |
10.1002/pi.6434 |
| IF(2015): |
3.213 |
| export: |
bibtex |
| pdf: |
<PolymerInt> |
The cover image is based on the Research Article A machine learning approach for the design of hyperbranched polymeric dispersing agents based on aliphatic polyesters for radiation-curable inks by Danny E.P. Vanpoucke et al., https://doi.org/10.1002/pi.6378. (The related paper can be found here.)

Cover Polymer International: Machine learning on small data sets, application on UV curable inks.
Permanent link to this article: https://dannyvanpoucke.be/paper2022_mldannycover-en/
| Authors: |
Danny E.P. Vanpoucke, Marie A.F. Delgove, Jules Stouten, Jurrie Noordijk, Nils De Vos, Kamiel Matthysen, Geert G.P. Deroover, Siamak Mehrkanoon, and Katrien V. Bernaerts |
| Journal: |
Polymer International 71(8), 966-975 (2022) |
| doi: |
10.1002/pi.6378 |
| IF(2021): |
3.213 |
| export: |
bibtex |
| pdf: |
<PI> (Open Access) (Cover Paper) |
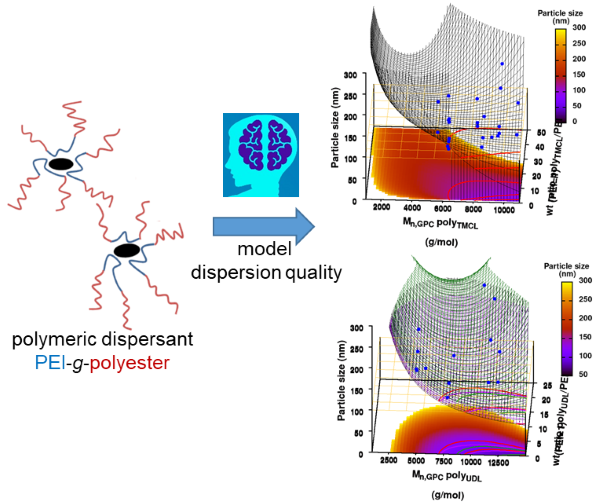 |
| Graphical Abstract:An ensemble based machine learning model for small datasets was used to predict the relationship between the dispersant structure and the pigment dispersion quality (particle size) for radiation curable formulations. |
Polymeric dispersing agents were prepared from aliphatic polyesters consisting of δ-undecalactone (UDL) and β,δ-trimethyl-ε-caprolactones (TMCL) as biobased monomers, which are polymerized in bulk via organocatalysts. Graft copolymers were obtained by coupling of the polyesters to poly(ethylene imine) (PEI) in the bulk without using solvents. Different parameters that influence the performance of the dispersing agents in pigment based UV-curable matrices were investigated: chemistry of the polyester (UDL or TMCL), weight ratio of polyester/PEI, molecular weight of the polyesters and of PEI. The performance of the dispersing agents was modelled using machine learning in order to increase the efficiency of the dispersant design. The resulting models were presented as analytical models for the individual polyesters and the synthesis conditions for optimal performing dispersing agents were indicated as a preference for high molecular weight polyesters and a polyester dependent maximum weight ratio polyester/PEI.
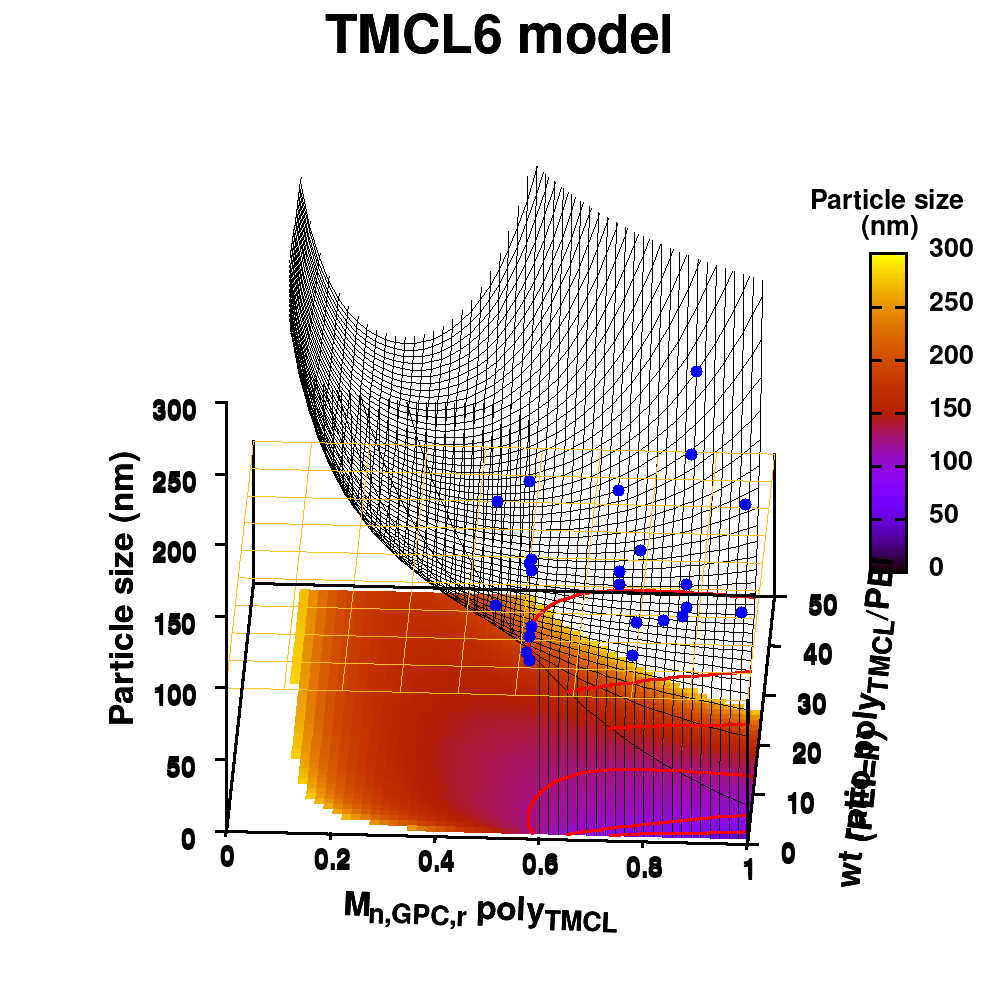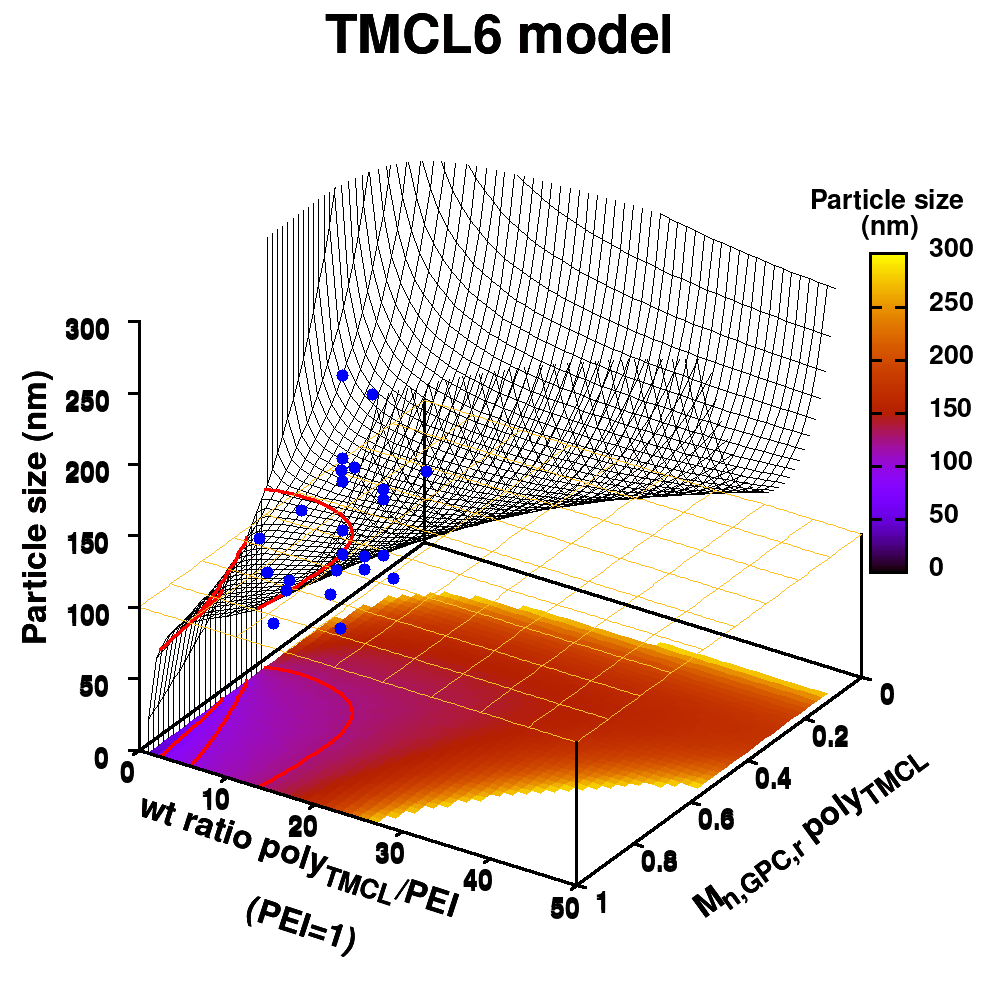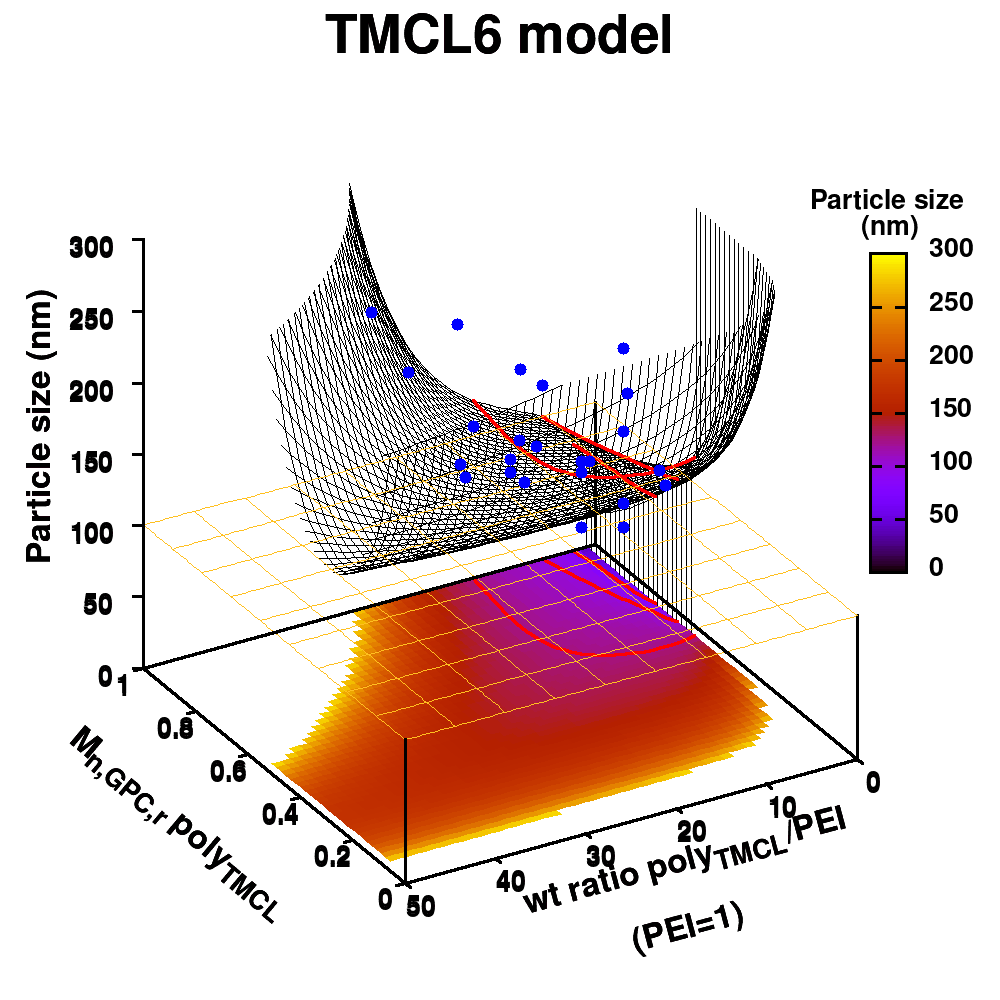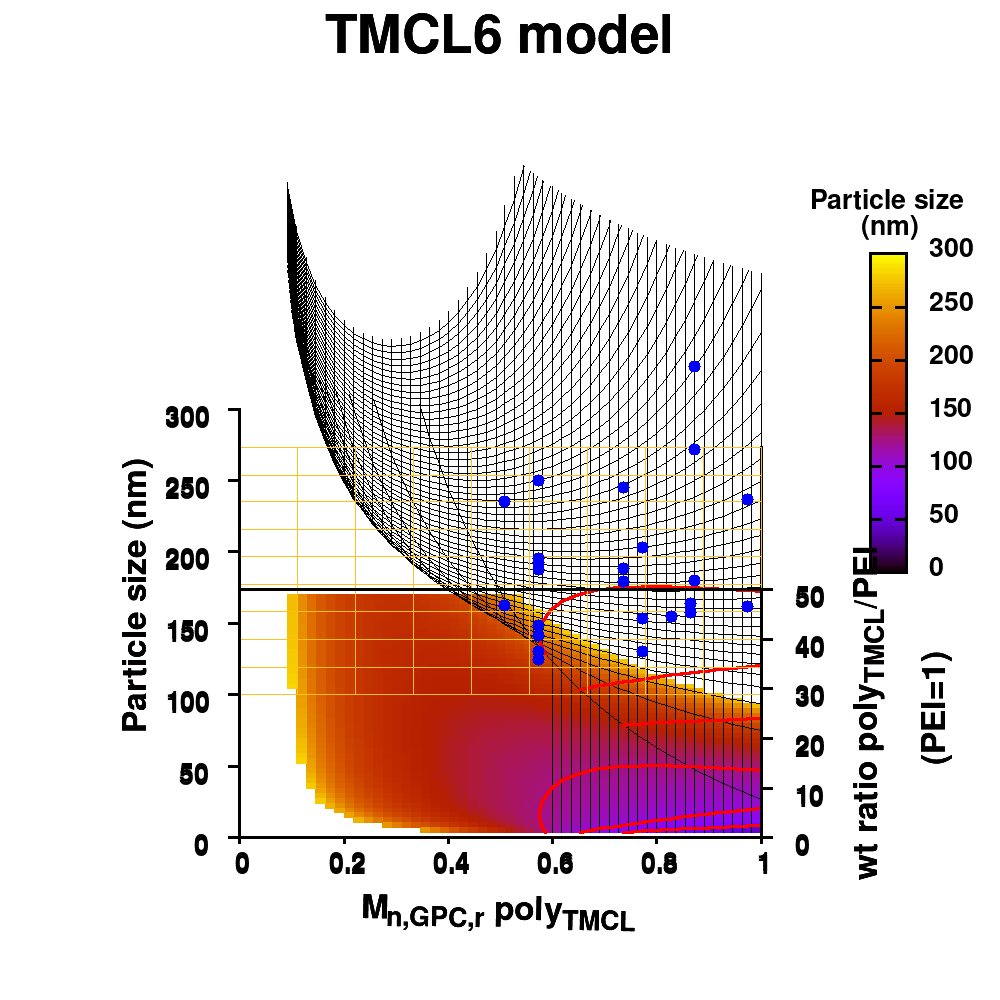
Animation of TMCL model 6
Permanent link to this article: https://dannyvanpoucke.be/paper-ml_inks_um-en/
| Authors: |
Kirill N. Boldyrev, Vadim S. Sedov, Danny E.P. Vanpoucke, Victor G. Ralchenko, & Boris N. Mavrin |
| Journal: |
Diam. Relat. Mater 126, 109049 (2022) |
| doi: |
10.1016/j.diamond.2022.109049 |
| IF(2020): |
3.315 |
| export: |
bibtex |
| pdf: |
<DRM> |
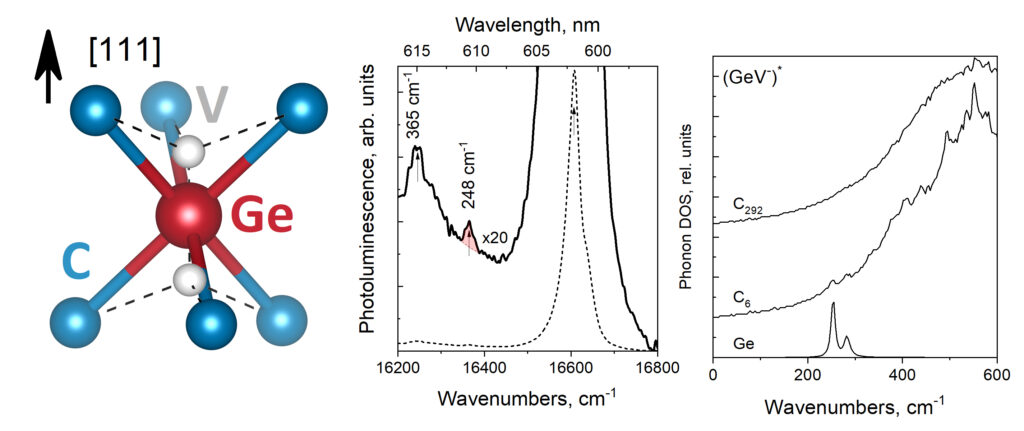 |
| Graphical Abstract: GeV split vacancy defect in diamond and the phonon modes near the ZPL. |
The vibrational behaviour of the germanium-vacancy (GeV) in diamond is studied through its photoluminescence spectrum and first-principles modeled partial phonon density of states. The former is measured in a region below 600 cm−1. The latter is calculated for the GeV center in its neutral, charged, and excited state. The photoluminescence spectrum presents a previously unobserved feature at 248 cm−1 in addition to the well-known peak at 365 cm−1. In our calculations, two localized modes, associated with the GeV center and six nearest carbon atoms (GeC6 cluster) are identified. These correspond to one vibration of the Ge ion along with the [111] orientation of the crystal and one perpendicular to this direction. We propose these modes to be assigned to the two features observed in the photoluminescence spectrum. The dependence of the energies of the localized modes on the GeV-center and their manifestation in experimental optical spectra is discussed.
Permanent link to this article: https://dannyvanpoucke.be/paper_gevpldft_vadim-en/








