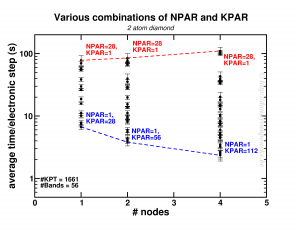
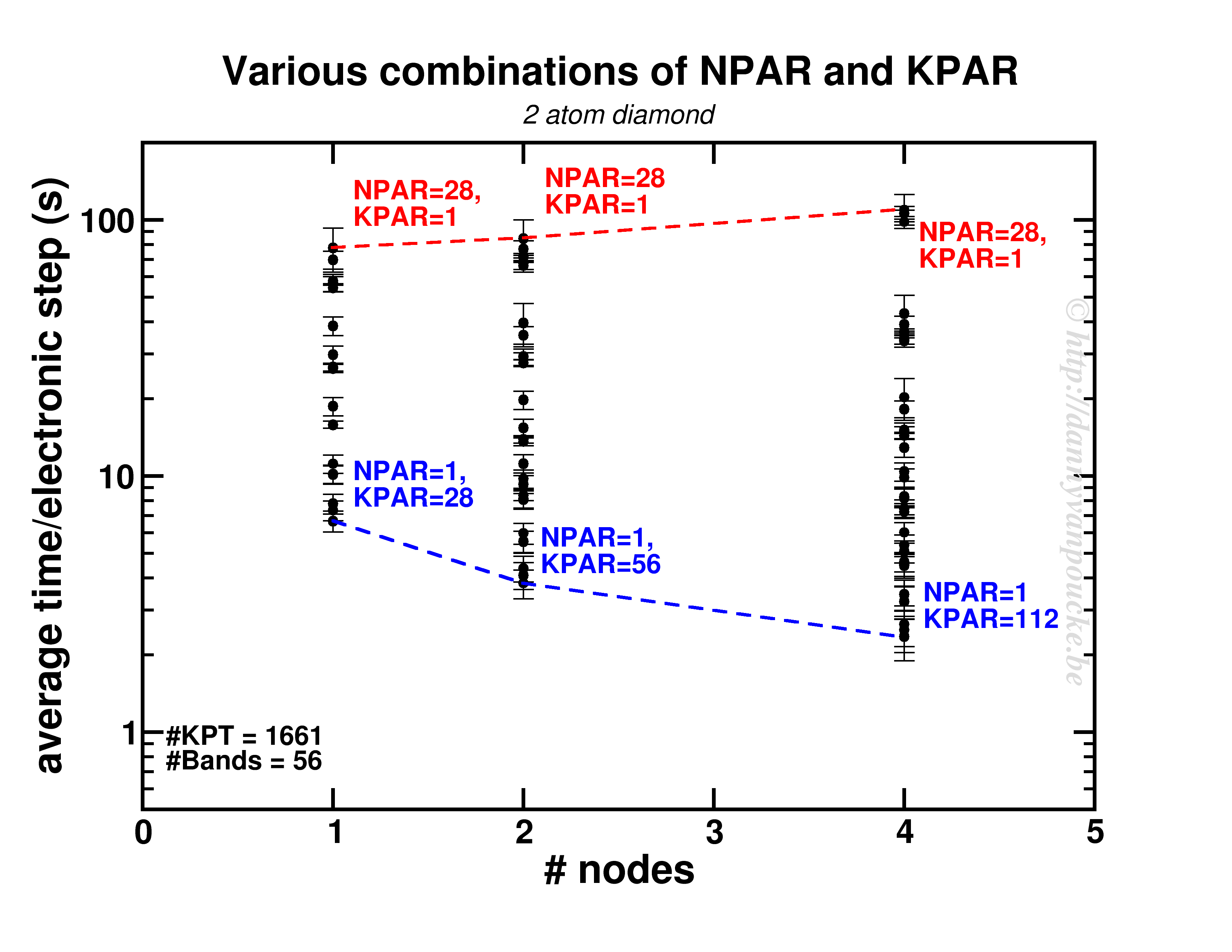
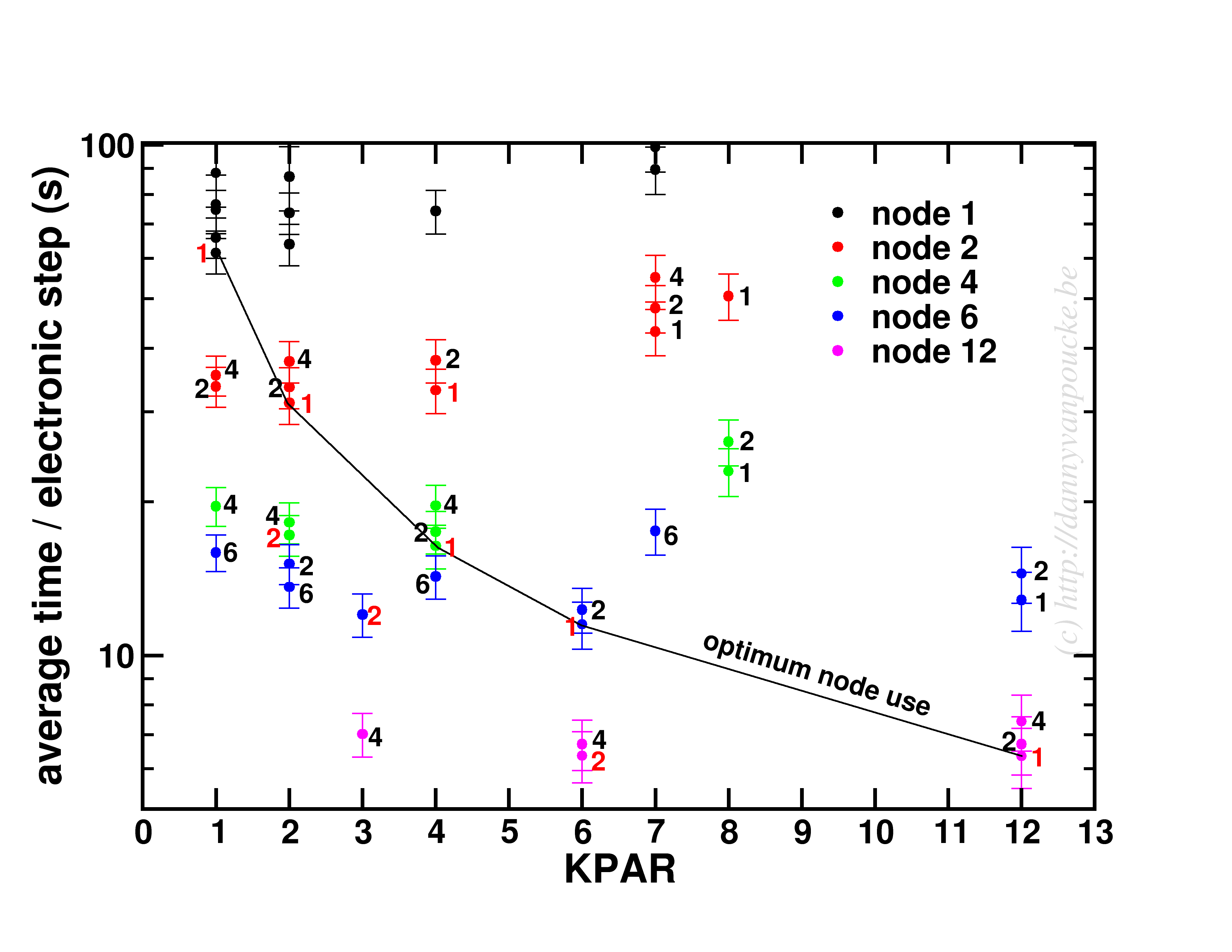
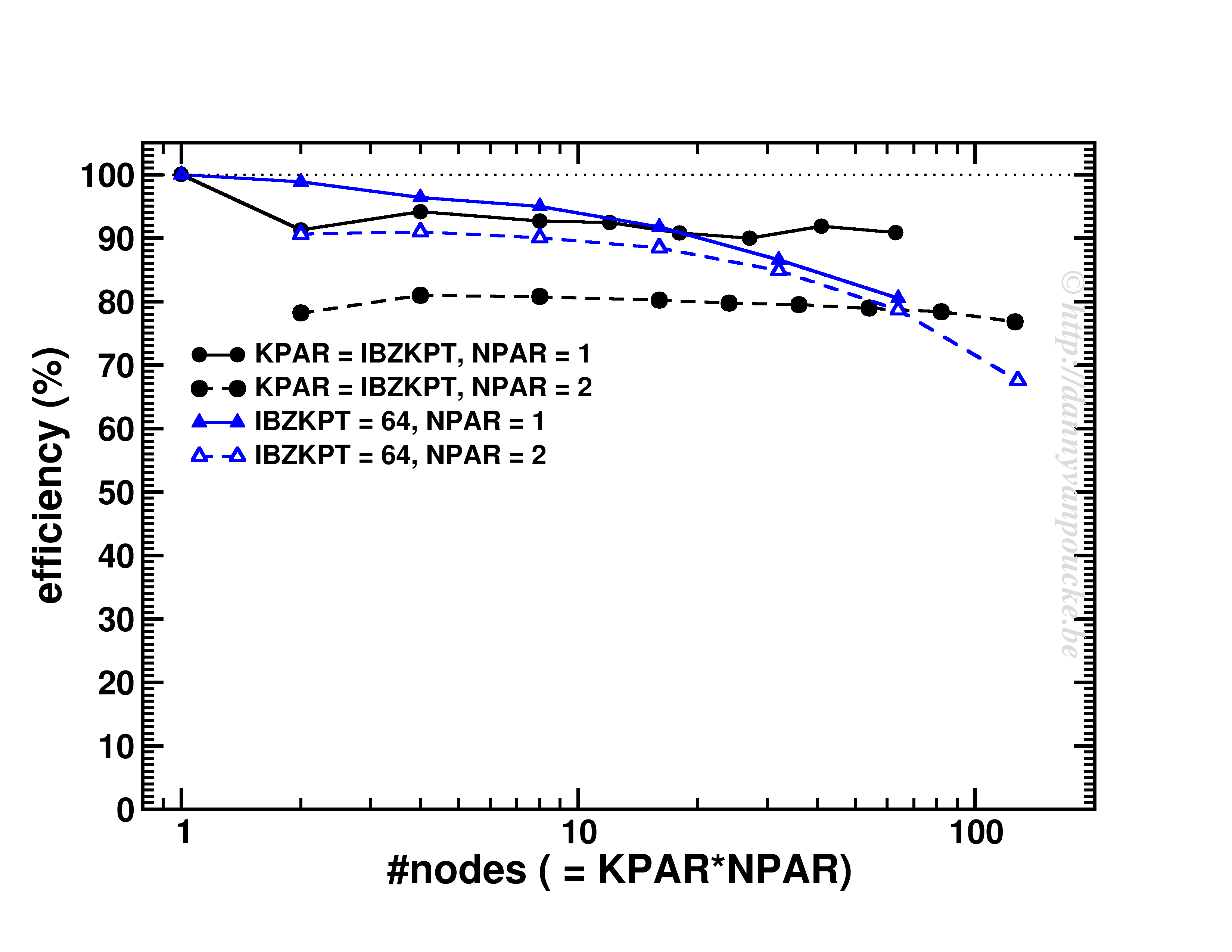
Happy New Year
2017 has come and gone. 2018 eagerly awaits getting acquainted. But first we look back one last time, trying to turn this into a old tradition. What have I done during the last year of some academic merit.
Publications: +4
-
Diam. Relat. Mater. 79, 60-69 (2017),
doi: 10.1016/j.diamond.2017.08.009 {IF(2016)=2.561} -
J. Alloys Compd. 729, 240-248 (2017),
doi: 10.1016/j.jallcom.2017.09.168 {IF(2016)=3.133} -
Phys. Chem. Chem. Phys. 19, 12414-12424 (2017),
doi: 10.1039/C7CP00998D {IF(2015)=4.449} -
J. Phys. Chem. C 121(14), 8014-8022 (2017),
doi: 10.1021/acs.jpcc.7b01491 {IF(2015)=4.509}
Completed refereeing tasks: +8
- The Journal of Physical Chemistry (2x)
- Journal of Physics: Condensed Matter (3x)
- Diamond and Related Materials (3x)
Conferences & workshops: +5 (Attended)
- Int. Conference on Diamond and Carbon Materials (DCM) 2017, Gothenburg, Sweden, September 3rd-7th, 2017 [oral presentation]
- Summerschool: “Upscaling techniques for mathematical models involving multiple scales”, Hasselt, Belgium, June 26th-29th, 2017 [poster presentation]
- VSC-user day, Brussels, Belgium, June 2nd, 2017 [poster presentation]
- E-MRS 2017 Spring Meeting, Strasbourg, France, May 22nd-26th, 2017 [1 oral + 2 poster presentations]
- SBDD XXII, Hasselt University, Belgium, March 8th-10th, 2017 [poster presentation]
PhD-students: +1
- Mohammadreza Hosseini (okt.-… ,Phd student physical chemistry, Tarbiat Modares University, Teheran, Iran)
Bachelor-students: +2
- Asja Vanwetswinkel (3rd Bach. Phys., Project: Applied Phonons)
- Giel Oosterbos (3rd Bach. Phys., Project: Ge-V complexes in Diamond)
Current size of HIVE:
- 48.5K lines of program (code: 70 %)
- ~70 files
- 45 (command line) options
Hive-STM program:
- 24 new users (making for a total of 300 users)