In the previous fortran tutorials, we learned the initial aspects of object oriented programming (OOP) in fortran 2003. And even though our agent-based opinion-dynamics-code is rather simple, it can quickly take several minutes for a single run of the program to finish. Two tools which quickly become of interest for codes that need more than a few minutes to run are: (1) a progress bar, to track the advance of the “slow” part of the code and prevent you from killing the program 5 seconds before it is to finish, and (2) a timer, allowing you to calculate the time needed to complete certain sections of code, and possibly make predictions of the expected total time of execution.
In this tutorial, we will focus on the progress bar. Since our (hypothetical) code is intended to run on High-Performance Computing (HPC) systems and is written in the fortran language, there generally is no (or no easy) access to GUI’s. So we need our progress bar class to run in a command line user interface. Furthermore, because it is such a widely useful tool we want to build it into a (shared) library (or dll in windows).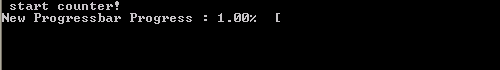
The progress bar class
What do we want out of our progress bar? It needs to be easy to use, flexible and smart enough to work nicely even for a lazy user. The output it should provide is formatted as follows: <string> <% progress> <text progress bar>, where the string is a custom character string provided by the user, while ‘%progress’ and ‘text progress bar’ both show the progress. The first shows the progress as an updating number (fine grained), while the second shows it visually as a growing bar (coarse grained).
[codesyntax lang=”fortran” lines=”normal” title=”TProgressBar Class” bookmarkname=”PBarClass” blockstate=”expanded” doclinks=”0″]
type, public :: TProgressBar
private
logical :: init
logical :: running
logical :: done
character(len=255) :: message
character(len=30) :: progressString
character(len=20) :: bar
real :: progress
contains
private
procedure,pass(this),public :: initialize
procedure,pass(this),public :: reset
procedure,pass(this),public :: run
procedure,pass(this),private:: printbar
procedure,pass(this),private:: updateBar
end type TProgressBar
[/codesyntax]
All properties of the class are private (data hiding), and only 3 procedures are available to the user: initialize, run and reset. The procedures, printbar and updatebar are private, because we intend the class to be smart enough to decide if a new print and/or update is required. The reset procedure is intended to reset all properties of the class. Although one might consider to make this procedure private as well, it may be useful to allow the user to reset a progress bar in mid progress.(The same goes for the initialize procedure.)
[codesyntax lang=”fortran” lines=”normal” title=”Run procedure of the TProgressBar class” blockstate=”expanded” bookmarkname=”RunProcedure” ]
subroutine run(this,pct,Ix,msg)
class(TProgressBar) :: this
real::pct
integer, intent(in), optional :: Ix
character(len=*),intent(in),optional :: msg
if (.not. this%init) call this%initialize(msg)
if (.not. this%done) then
this%running=.true.
this%progress=pct
call this%updateBar(Ix)
call this%printbar()
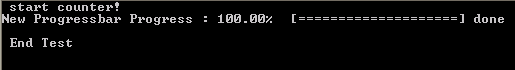
if (abs(pct-100.0)<1.0E-6) then
this%done=.true.
write(*,'(A6)') "] done"
end if
end if
end subroutine run
[/codesyntax]
In practice, the run procedure is the heart of the class, and the only procedure needed in most applications. It takes 3 parameters: The progress (pct), the number of digits to print of pct (Ix),and the <string> message (msg). The later two parameters are even optional, since msg may already have been provided if the initialize procedure was called by the user. If the class was not yet initialized it will be done at the start of the procedure. And while the progress bar has not yet reached 100% (within 1 millionth of a %) updates and prints of the bar are performed. Using a set of Boolean properties (init, running, done), the class keeps track of its status. The update and print procedures just do this: update the progress bar data and print the progress bar. To print the progress bar time and time again on the same line, we need to make use of the carriage return character (character 13 of the ASCII table):
write(*,trim(fm), advance='NO') achar(13), trim(this%message),trim(adjustl(this%progressString)),'%','[',trim(adjustl(this%bar))
The advance=’NO‘ option prevents the write statement to move to the next line. This can sometimes have the unwanted side-effect that the write statement above does not appear on the screen. To force this, we can use the fortran 2003 statement flush(OUTPUT_UNIT), where “OUTPUT_UNIT” is a constant defined in the intrinsic fortran 2003 module iso_fortran_env. For older versions of fortran, several compilers provided a (non standard) flush subroutine that could be called to perform the same action. As such, we now have our class ready to be used. The only thing left to do is to turn it into a dll or shared library.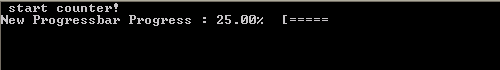
How to create a library and use it
There are two types of libraries: static and dynamic.
Static libraries are used to provide access to functions/subroutines at compile time to the library user. These functions/subroutines are then included in the executable that is being build. In linux environments these will have the extension “.a”, with the .a referring to archive. In a windows environment the extension is “.lib”, for library.
Dynamic libraries are used to provide access to functions/subroutines at run time. In contrast to static libraries, the functions are not included in the executable, making it smaller in size. In linux environments these will have the extension “.so”, with the .so referring to shared object. In a windows environment the extension is “.dll”, for dynamically linked library.
In contrast to C/C++, there is relatively little information to be found on the implementation and use of libraries in fortran. This may be the reason why many available fortran-“libraries” are not really libraries, in the sense meant here. Instead they are just one or more files of fortran code shared by their author(s), and there is nothing wrong with that. These files can then be compiled and used as any other module file.
So how do we create a library from our Progressbar class? Standard examples start from a set of procedures one wants to put in a library. These procedures are put into a .f or .f90 file. Although they are not put into a module (probably due to the idea of having compatibility with fortran 77) which is required for our class, this is not really an issue. The same goes for the .f03 or .f2003 extension for our file containing a fortran 2003 class. To have access to our class and its procedures in our test program, we just need to add the use progressbarsmodule clause. This is because our procedures and class are incorporated in a module (in contrast to the standard examples). Some of the examples I found online also include compiler dependent pragmas to export and import procedures from a dll. Since I am using gfortran+CB for development, and ifort for creating production code, I prefer to avoid such approaches since it hampers workflow and introduces another possible source of bugs.
The compiler setups I present below should not be considered perfect, exhaustive or fool-proof, they are just the ones that work fine for me. I am, however, always very interested in hearing other approaches and fixes in the comments.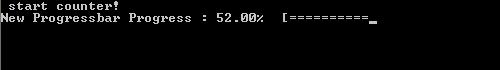
Windows
The windows approach is very easy. We let Code::Blocks do all the hard work.
shared library: PBar.dll
Creating the dll : Start a new project, and select the option “Fortran DLL“. Follow the instructions, which are similar to the setup of a standard fortran executable. Modify/replace/add the fortran source you wish to include into your library and build your code (you can not run it since it is a library).
Creating a user program : The program in which you will be using the dll is setup in the usual way. And to get the compilation running smoothly the following steps are required:
- Add the use myspecificdllmodule clause where needed, with myspecificdllmodule the name of the module included in the dll you wish to use at that specific point.
- If there are modules included in the dll, the *.mod files need to be present for the compiler to access upon compilation of the user program. (Which results in a limitation with regard to distribution of the dll.)
- Add the library to the linker settings of the program (project>build options>linker settings), and then add the .dll file.
- Upon running the program you only need the program executable and the dll.
static library
The entire setup is the same as for the shared library. This time, however, choose the “Fortran Library” option instead of Fortran dll. As the static library is included in the executable, there is no need to ship it with the executable, as is the case for the dll.
Unix
For the unix approach we will be working on the command line, using the intel compiler, since this compiler is often installed at HPC infrastructures.
static library: PBar.a
After having created the appropriate fortran files you wish to include in your library (in our example this is always a single file: PBar.f03, but for multiple files you just need to replace PBar.f03 with the list of files of interest.)
- Create the object files:
ifort -fpic -c -free -Tf Pbar.f03
Where -fpic tells the compiler to generate position independent code, typical for use in a shared object/library, while -c tells the compiler to create an object file. The -free and -Tf compiler options are there to convince the compiler that the f03 file is actual fortran code to compile and that it is free format.
- Use the GNU ar tool to combine the object files into a library:
ar rc PBarlib.a PBar.o
- Compile the program with the library
ifort TestProgram.f90 PBarlib.a -o TestProgram.exe
Note that also here the .mod file of our Progressbarsmodule needs to be present for the compilation to be successful.
shared library: PBar.so
For the shared library the approach does not differ that much.
- Create the object files:
ifort -fpic -c -free -Tf Pbar.f03
In this case the fpic option is not optional in contrast to the static library above. The other options are the same as above.
- Compile the object files into a shared library:
ifort -shared PBar.o -o libPBar.so
The compiler option -shared creates a shared library, while the -o option allows us to set the name of the library.
- Compile the program with the library
ifort TestProgram.f90 libPBar.so -o TestProgram.exe
Note that also here the .mod file of our Progressbarsmodule needs to be present for the compilation to be successful. To run the program you also need to add the location of the library file libPBar.so to the environment variable LD_LIBRARY_PATH
One small pickle
HPC systems may perform extensive buffering of data before output, to increase the efficiency of the machine (disk-writes are the slowest memory access option)…and as a result this can sometimes overrule our flush command. The progressbar in turn will not show much progress until it is actually finished, at which point the entire bar will be shown at once. There are options to force the infrastructure not to use this buffering (and the system administrators in general will not appreciate this), for example by setting the compiler flag -assume nobuffered_stdout. So the best solution for HPC applications will be the construction of a slightly modified progress bar, where the carriage return is not used.
Special thanks also to the people of stack-exchange for clarifying some of the issues with the modules.
Source files for the class and test-program can be downloaded here.









 aka
aka  . In response to the creation of
. In response to the creation of