Most commented posts
- Start to Fortran — 1 comment
- Phonons: shake those atoms — 3 comments


Permanent link to this article: https://dannyvanpoucke.be/book-chapter-mofs-en/
Jan 09 2018
Over the last weekend, two serious cyber security issues were hot news: Meltdown and Spectre [more links, and links](not to be mistaken for a title of a bond-movie). As a result, also academic HPC centers went into overdrive installing patches as fast as possible. The news of the two security issues went hand-in-hand with quite a few belittling comments toward the chip-designers ignoring the fact that no-one (including those complaining now) discovered the problem for over decade. Of course there was also the usual scare-mongering (cyber-criminals will hack our devices by next Monday, because hacks using these bugs are now immediately becoming their default tools etc.) typical since the beginning of the 21st century…but now it is time to return back to reality.
One of the big users on scientific HPC installations is the VASP program(an example), aimed at the quantum mechanical simulation of materials, and a program central to my own work. Due to an serendipitous coincidence of a annoyingly hard to converge job I had the opportunity to see the impact of the Meltdown and Spectre patches on the performance of VASP: 16% performance loss (within the range of the expected 10-50% performance loss for high performance applications [1][2][3]).
The case:
The calculation took several runs of about 10 electronic steps (of each about 5-6 h wall-time, about 2.54 years of CPU-time per run) . The relative average time is shown below (error-bars are the standard deviation of the times within a single run). As the final step takes about 50% longer it is treated separately. As you can see, the variation in time between different electronic steps is rather small (even running on a different cluster only changes the time by a few %). The impact of the Meltdown/Spectre patch gives a significant impact.
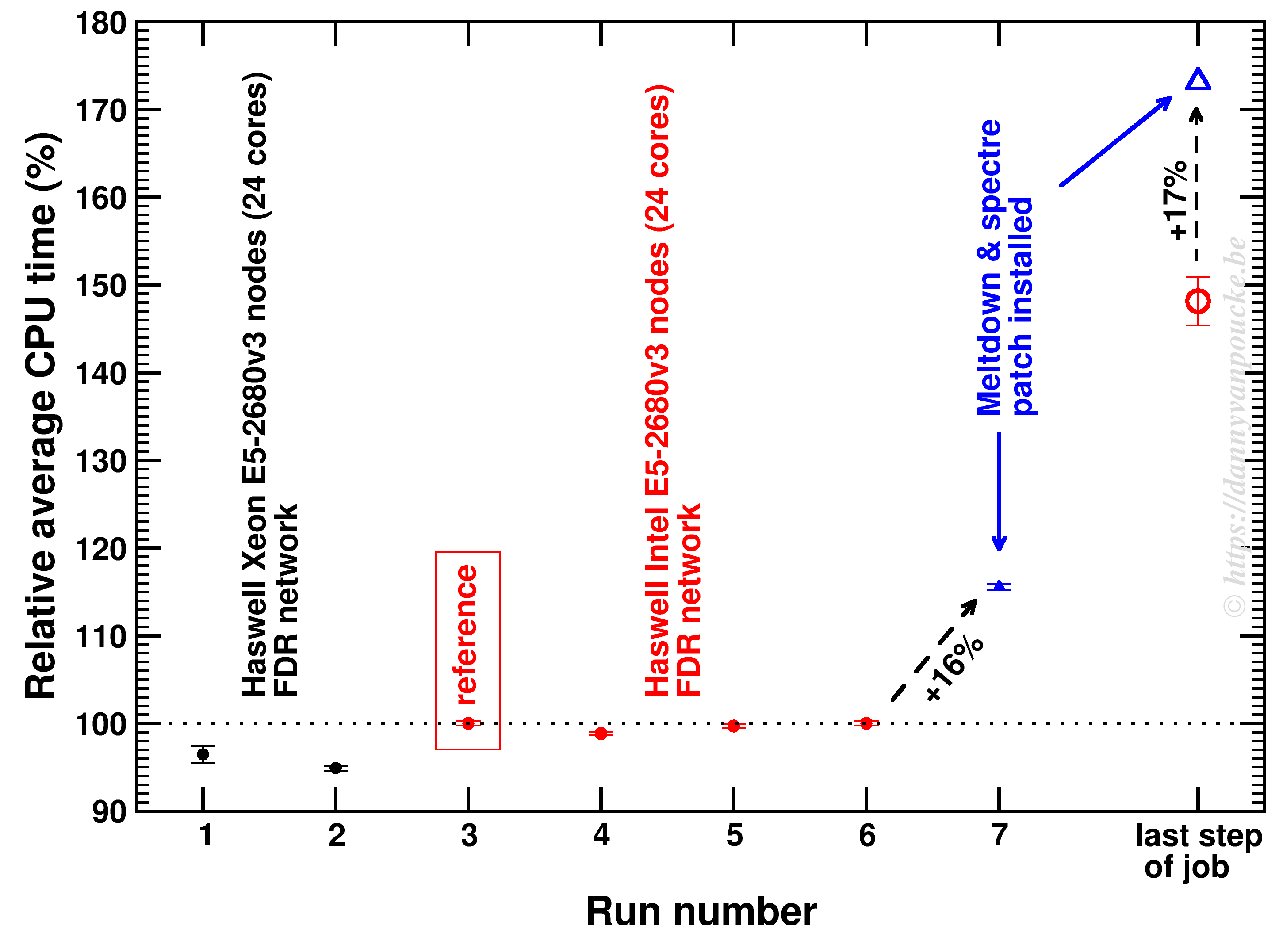
Impact of Meltdown/Spectre patch on VASP performance for a 336 core MPI job.
The HPC team is currently looking into possible workarounds that could (partially) alleviate the problem. VASP itself is rather little I/O intensive, and a first check by the HPC team points toward MPI (the parallelisation framework required for multi-node jobs) being ‘a’ if not ‘the’ culprit. This means that also an impact on other multi-node programs is to be expected. On the bright side, finding a workaround for MPI would be beneficial for all of them as well.
So far, tests I performed with the HPC team not shown any improvements (recompiling VASP didn’t help, nor an MPI related fix). Let’s keep our fingers crossed, and hope the future brings insight and a solution.
Permanent link to this article: https://dannyvanpoucke.be/spectre-and-meltdown-en/
Jan 01 2018
2017 has come and gone. 2018 eagerly awaits getting acquainted. But first we look back one last time, trying to turn this into a old tradition. What have I done during the last year of some academic merit.
Publications: +4
Completed refereeing tasks: +8
Conferences & workshops: +5 (Attended)
PhD-students: +1
Bachelor-students: +2
Current size of HIVE:
Hive-STM program:
Permanent link to this article: https://dannyvanpoucke.be/review-of-2017-en/
Nov 12 2017

Simulated STM image of the Pt-induced nanowires on the Ge(001) surface. Green discs indicate the atomic positions of the bulk-Ge atoms; red: Pt atoms embedded in the top surface layers; yellow: Ge atoms forming the nanowire observed by STM.
Ten years ago, I was happily modeling Pt nanowires on Ge(001) during my first Ph.D. at the university of Twente. As a member of the Computational Materials Science group, I also was lucky to have good and open contact with the experimental research group of Prof. Zandvliet, whom was growing these nanowires. In this environment, I learned there is a big difference between what is easy in experiment and what is easy in computational research. It also taught me to find a common ground which is “easy” for both (Scanning tunneling microscopy (STM) images in this specific case).
During this 4-year project, I quickly came to the conclusion that the nanowires could not be formed by Pt atoms, but that it needed to be Ge atoms instead. Although the simulated STM images were very convincing, it was really hard to overcome the experimental intuition…and experiments which seemed to contradict this picture (doi: 10.1016/j.susc.2006.07.055 ). As a result, I spend a lot of time learning about the practical aspects of the experiments (an STM tip is a complicated thing) and trying to extract every possible piece of information published and unpublished. Especially the latter provided important support. The “ugly”(=not good for publishing) experimental pictures tended to be real treasures from my computational point of view. Of course, much time was spent on tweaking the computational model to get a perfect match with experiments (e.g. the 4×1 periodicity), and trying to reproduce experiments seemingly supporting the “Ge-nanowire” model (e.g. simulation of CO adsorption and identification of the path along the wire the molecule follows.).
In contrast to my optimism at the end of my first year (I believed all modeling could be finished before my second year ended), the modeling work ended up being a very complex exercise, taking 4 years of research. Now I am happy that I was wrong, as the final result ended up being very robust and became “The model for Pt induced nanowires on Ge(001)“.
Upon doing a review article on this field five years after my Ph.D. I was amazed (and happy) to see my model still stood. Even more, there had been complex experimental studies (doi: 10.1103/PhysRevB.85.245438) which even seemed to support the model I proposed. However, these experiments were stil making an indirect comparison. A direct comparison supporting the Ge nature of the nanowires was still missing…until recently.
In a recent paper in Phys. Rev. B (doi: 10.1103/PhysRevB.96.155415) a Japanese-Turkish collaboration succeeded in identifying the nanowire atoms as Ge atoms. They did this using an Atomic Force Microscope (AFM) and a sample of Pt induced nanowires, in which some of the nanowire atoms were replaced by Sn atoms. The experiment rather simple in idea (execution however requires rather advanced skills): compare the forces experienced by the AFM when measuring the Sn atom, the chain atoms and the surface atoms. The Sn atoms are easily recognized, while the surface is known to consist of Ge atoms. If the relative force of the chain atom is the same as that of the surface atoms, then the chain consists of Ge atoms, while if the force is different, the chain consists of Pt atoms.
*small drum-roll*
And they found the result to be the same.
Yes, after nearly 10 years since my first publication on the subject, there finally is experimental proof that the Pt nanowires on Ge(001) consist of Ge atoms. Seeing this paper made me one happy computational scientist. For me it shows the power of computational research, and provides an argument why one should not be shy to push calculations to their limit. The computational cost may be high, but at least one is performing relevant work. And of course, never forget, the most seemingly easy looking experiments are usually not easy at all, so as a computational materials scientist you should not take them for granted, but let those experimentalists know how much you appreciate their work and effort.
Permanent link to this article: https://dannyvanpoucke.be/slow-science-ptnw-en/
Oct 26 2017
One of the new features provided by Elsevier upon publication is the creation of audioslides. This is a kind of short presentation of the publication by one of the authors. I have been itching to try this since our publication on the neutral C-vancancy was published. The interface is quite intuitive, although the adobe flash tend to have a hard time finding the microphone. However, once it succeeds, things go quite smoothly. The resolution of the slides is a bit low, which is unfortunate (but this is only for the small-scale version, the large-scale version is quite nice as you can see in the link below). Maybe I’ll make a high resolution version video and put it on Youtube, later.
The result is available here (since the embedding doesn’t play nicely with WP).
And a video version can be found here.
Permanent link to this article: https://dannyvanpoucke.be/audioslides-tryout-en/
Sep 23 2017
| Authors: | Danny E. P. Vanpoucke and Ken Haenen |
| Journal: | Diam. Relat. Mater 79, 60-69 (2017) |
| doi: | 10.1016/j.diamond.2017.08.009 |
| IF(2017): | 2.232 |
| export: | bibtex |
| pdf: | <DiamRelatMater> |
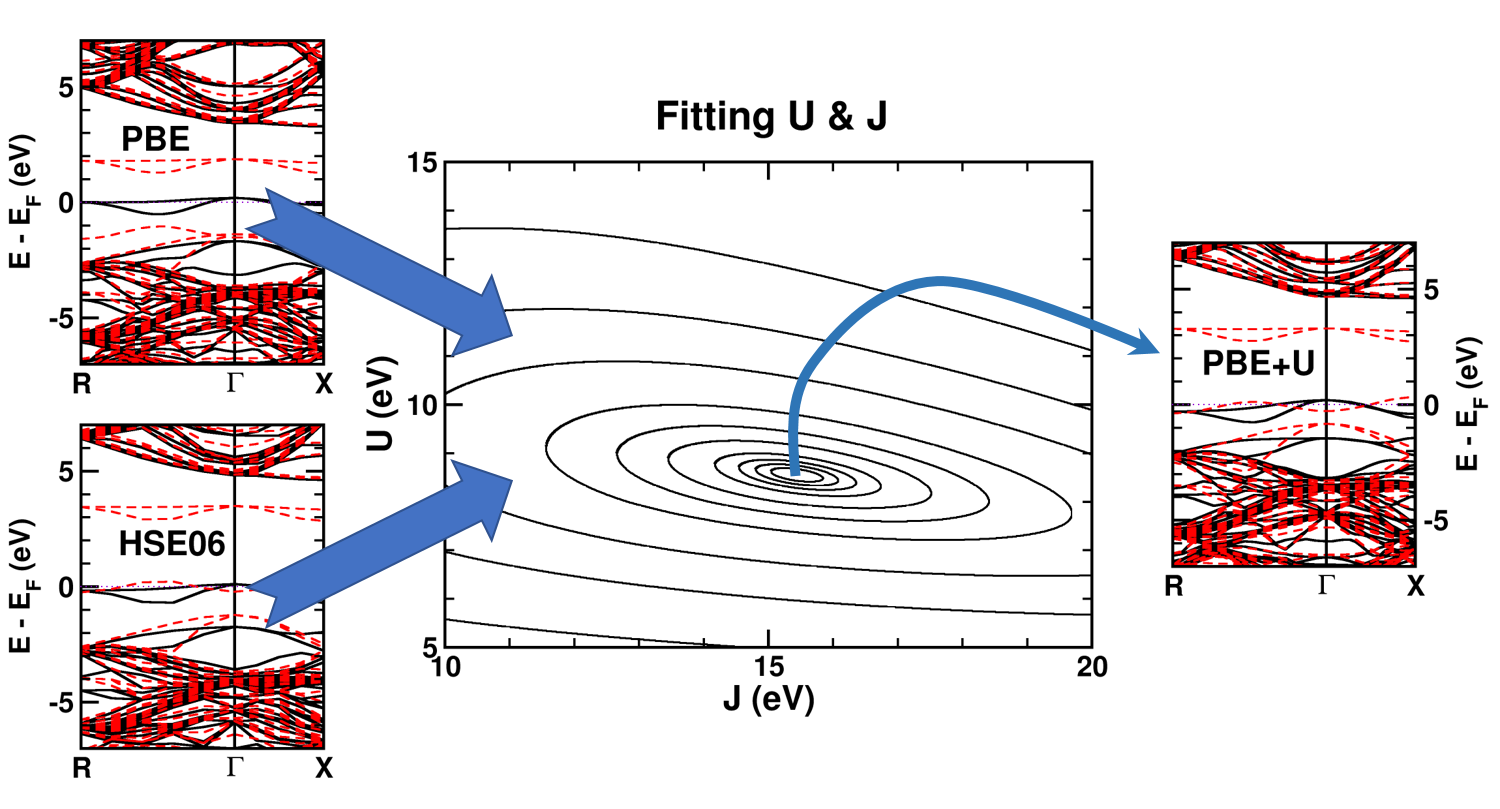 |
| Graphical Abstract: Combining a scan over possible values for U and J with reference electronic structures obtained using the hybrid functional HSE06, DFT+U can be fit to provide hybrid functional quality electronic structures at the cost of DFT calculations. |
The neutral C-vacancy is investigated using density functional theory calculations. We show that local functionals, such as PBE, can predict the correct stability order of the different spin states, and that the success of this prediction is related to the accurate description of the local magnetic configuration. Despite the correct prediction of the stability order, the PBE functional still fails predicting the defect states correctly. Introduction of a fraction of exact exchange, as is done in hybrid functionals such as HSE06, remedies this failure, but at a steep computational cost. Since the defect states are strongly localized, the introduction of additional on site Coulomb and exchange interactions, through the DFT+U method, is shown to resolve the failure as well, but at a much lower computational cost. In this work, we present optimized U and J parameters for DFT+U calculations, allowing for the accurate prediction of defect states in defective
diamond. Using the PBE optimized atomic structure and the HSE06 optimized electronic structure as reference, a pair of on-site Coulomb and exchange parameters (U,J) are fitted for DFT+U studies of defects in diamond.
Poster-presentation: here
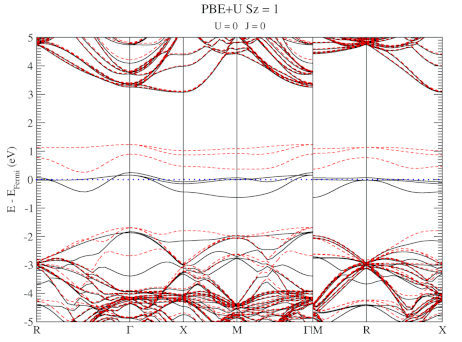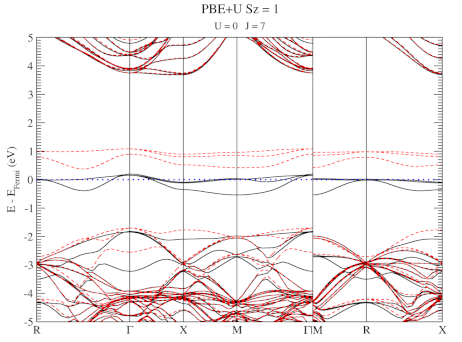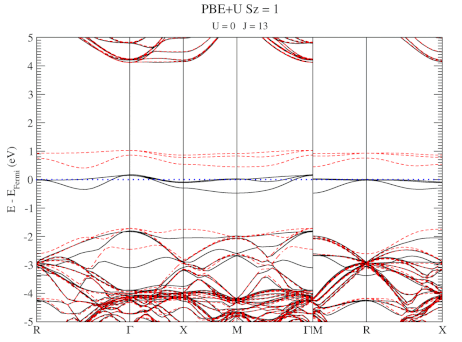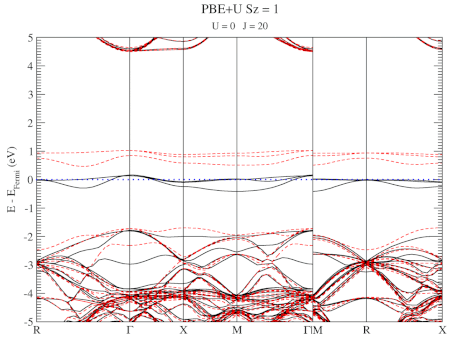
DFT+U series (varying J) for a specific spin state of the C-vacancy defect.
Permanent link to this article: https://dannyvanpoucke.be/paperdrm-diamond-dftu-en/
Sep 23 2017
| Authors: | Seyyed Amin Rounaghi, Danny E.P. Vanpoucke, Hossein Eshghi, Sergio Scudino, Elaheh Esmaeili, Steffen Oswald and Jürgen Eckert |
| Journal: | J. Alloys Compd. 729, 240-248 (2017) |
| doi: | 10.1016/j.jallcom.2017.09.168 |
| IF(2017): | 3.779 |
| export: | bibtex |
| pdf: | <J.Alloys Compd.> |
 |
| Graphical Abstract: Evolution of the end products as function of Al and N content during ball-milling synthesis of AlN. |
A versatile ball milling process was employed for the synthesis of hexagonal aluminum nitride (h-AlN) through the reaction of metallic aluminum with melamine. A combined experimental and theoretical study was carried out to evaluate the synthesized products. Milling intermediates and products were fully characterized via various techniques including XRD, FTIR, XPS, Raman and TEM. Moreover, a Boltzmann distribution model was proposed to investigate the effect of milling energy and reactant ratios on the thermodynamic stability and the proportion of different milling products. According to the results, the reaction mechanism and milling products were significantly influenced by the reactant ratio. The optimized condition for AlN synthesis was found to be at Al/M molar ratio of 6, where the final products were consisted of nanostructured AlN with average crystallite size of 11 nm and non-crystalline heterogeneous carbon.
Permanent link to this article: https://dannyvanpoucke.be/paperdoped-nitrides-iii-en/
Aug 29 2017
As a computational materials scientist with a main research interest in the ab initio simulation of materials, computational resources are the life-blood of my research. Over the last decade, I have seen my resource usage grow from less than 100.000 CPU hours per year to several million CPU-hours per year. To satisfy this need for computational resources I have to make use of HPC facilities, like the TIER-2 machines available at the Flemish universities and the Flemish TIER-1 supercomputer, currently hosted at KU Leuven. At the international level, computational scientists have access to so called TIER-0 machines, something I no doubt will make use of in the future. Before I continue, let me first explain a little what this TIER-X business actually means.
The TIER-X notation is used to give an indication of the size of the computer/supercomputer indicated. There are 4 sizes:
This is roughly the status of what is available today for Flemish scientists at various levels. With the constantly growing demand for more processing power, the European union, in name of EuroHPC, has decided in march of this year, that Europe will host two Exa-scale computers. These machines will have a peak performance of at least 1 EFLOPS, or 1000 PFLOPS. These machines are expected to be build by 2024-2025. In June, Belgium signed up to EuroHPC as the eighth country participating, in addition, to the initial 7 countries (Germany, France, Spain, Portugal, Italy, Luxemburg and The Netherlands).
This is very good news for all involved in computational research in Flanders. There is the plan to build these machines, there is a deadline, …there just isn’t an idea of what these machines should look like (except: they will be big, massively power consuming and have a target peak performance). To get an idea what users expect of such a machine, Tier-1 and HPC users have been asked to put forward requests/suggestions of what they want.
From my user personal experience, and extrapolating from my own usage I see myself easily using 20 million hours of CPU time each year by the time these Exa-scale machines are build. Leading a computational group would multiply this value. And then we are talking about simple production purpose calculations for “standard” problems.
The claim that an Exa-scale scale machine runs 1000x faster than a peta-scale machine, is not entirely justified, at least not for the software I am generally encountering. As software seldom scales linearly, the speed-gain from Exa-scale machinery mainly comes from the ability to perform many more calculations in parallel. (There are some exceptions which will gain within the single job area, but this type of jobs is limited.) Within my own field, quantum mechanical calculation of the electronic structure of periodic atomic systems, the all required resources tend to grow with growth of the problem size. As such, a larger system (=more atoms) requires more CPU-time, but also more memory. This means that compute nodes with many cores are welcome and desired, but these cores need the associated memory. Doubling the cores would require the memory on a node to be doubled as well. Communication between the nodes should be fast as well, as this will be the main limiting factor on the scaling performance. If all this is implemented well, then the time to solution of a project (not a single calculation) will improve significantly with access to Exa-scale resources. The factor will not be 100x from a Pflops system, but could be much better than 10x. This factor 10 also takes into account that projects will have access to much more demanding calculations as a default (Hybrid functional structure optimization instead of simple density functional theory structure optimization, which is ~1000x cheaper for plane wave methods but is less accurate).
At this scale, parallelism is very important, and implementing this into a program is far from a trivial task. As most physicists/mathematicians/chemists/engineers may have the skills for writing scientifically sound software, we are not computer-scientists and our available time and skills are limited in this regard. For this reason, it will become more important for the HPC-facility to provide parallelization of software as a service. I.e. have a group of highly skilled computer scientists available to assist or even perform this task.
Next to having the best implementation of software available, it should also be possible to get access to these machines. This should not be limited to a happy few through a peer review process which just wastes human research potential. Instead access to these should be a mix of guaranteed access and peer review.
What could an HPC user as myself do to contribute to the success of EuroHPC? This is rather simple, run the machine as a pilot user (I have experience on most of the TIER-2 clusters of Ghent University and both Flemish Tier-1 machines. I successfully crashed the programs I am using by pushing them beyond their limits during pilot testing, and ran into rather unfortunate issues. 🙂 That is the job of a pilot user, use the machine/software in unexpected ways, such that this can be resolved/fixed by the time the bulk of the users get access.) and perform peer review of the lager specialized projects.
Now the only thing left to do is wait. Wait for the Exa-scale supercomputers to be build…7 years to go…about 92 node-days on Breniac…a starting grant…one long weekend of calculations.
For simplicity I use the term CPU to indicate a single compute core, even though technically, nowadays a single CPU will contain multiple cores (desktop/laptop: 2-8 cores, HPC-compute node: 2-20 cores / CPU (or more) ). This to make comparison a bit more easy.
Furthermore, modern computer systems start more and more to rely on GPU performance as well, which is also a possible road toward Exa-scale computing.
Permanent link to this article: https://dannyvanpoucke.be/exa-scale-computing/
Aug 05 2017
 Today is a joyful day, as I am getting married to my lovely girlfriend, my partner in crime, the mother of our son, the stars of my night-sky: my wife.
Today is a joyful day, as I am getting married to my lovely girlfriend, my partner in crime, the mother of our son, the stars of my night-sky: my wife.
In February, we silently snuck out and got married (just the two of us and our son). We even had a small honeymoon/pilgrimage to Enschede, the place where it all began. We visited the town and the university campus, reliving old memories of those first days.
Today, we have the ring-ceremony and wedding party to share our happiness with our family and friends. As we are having the celebration at a local castle, Sylvia also designed a special coat of arms for our little family:

Permanent link to this article: https://dannyvanpoucke.be/just-married/
Jul 29 2017
Computational as a third pillar of science (next to experimental and theoretical) is steadily developing in many fields of science. Even some fields you would less expect it, such as sociology or psychology. In other fields such as physics, chemistry or biology it is much more widespread, with people pushing the boundaries of what is possible. Larger facilities provide access to larger problems to tackle. If a computational physicist is asked if larger infrastructures would not become too big, he’ll just shrug and reply: “Don’t worry, we will easily fill it up, even a machine 1000x larger than that.” An example is given by a pair of physicists who recently published their atomic scale study of the HIV-1 virus. Their simulation of a model containing more than 64 million atoms used force fields, making the simulation orders of magnitude cheaper than quantum mechanical calculations. Despite this enormous speedup, their simulation of 1.2 µs out of the life of an HIV-1 virus (actually it was only the outer skin of the virus, the inside was left empty) still took about 150 days on 3880 nodes of 16 cores on the Titan super computer of Oak Ridge National Laboratory (think about 25 512 years on your own computer).
In Flanders, scientist can make use of the TIER-1 facilities provided by the Flemish Super Computer (VSC). The first Tier-1 machine was installed and hosted at Ghent University. At the end of it’s life cycle the new Tier-1 machine (Breniac) was installed and is hosted at KULeuven. Although our Tier-1 supercomputer is rather modest compared to the Oak Ridge supercomputer (The HIV-1 calculation I mentioned earlier would require 1.5 years of full time calculations on the entire machine!) it allows Flemish scientists (including myself) to do things which are not possible on personal desktops or local clusters. I have been lucky, as all my applications for calculation time were successful (granting me between 1.5 and 2.5 million hours of CPU time every year). With the installment of the new supercomputer accounting of the requested resources has become fully integrated and automated. Several commands are available which provide accounting information, of which mam-balance is the most important one, as it tells how much credits are still available. However, if you are running many calculations you may want to know how many resources you are actually asking and using in real-time. For this reason, I wrote a small bash-script that collects the number of requested and used resources for running jobs:
[codesyntax lang="bash" lines="normal" blockstate="collapsed"]
#small script to collect TIER-1 usage and requested resources
#!/bin/bash
echo "(c) dannyvanpoucke.be"
echo " _____"
echo " \0.0/"
echo " ( o )"
echo " ^-^ "
echo " Collecting Tier-1 requested/used resources: Breniac "
echo "-----------------------------------------------------"
#step one running resources
RCRrt="0"
RCRwt="0"
tnodes="0"
numl=`qstat -n | grep " R " | wc -l`
for (( i=1 ; i<= $numl ; i++ )); do
lin=`qstat -n | grep " R " | head -$i | tail -1`
nodes=`echo $lin | awk '{print $6}'`
tnodes=$( echo "$tnodes + $nodes" | bc )
lin=`echo $lin | sed 's/:/\ /g'`
rth=`echo $lin | awk '{print $13}'`
rtm=`echo $lin | awk '{print $14}'`
rts=`echo $lin | awk '{print $15}'`
wth=`echo $lin | awk '{print $9}'`
wtm=`echo $lin | awk '{print $10}'`
wts=`echo $lin | awk '{print $11}'`
rtuse=$( bc -l << EOF
scale=4
$nodes * (($rth + ( $rtm + ( $rts / 60.0000 ) )/60.0000)/24.00)
EOF
)
wtuse=$( bc -l << EOF
scale=4
$nodes * (($wth + ( $wtm + ( $wts / 60.0000 ) )/60.0000)/24.00)
EOF
)
RCRrt=$( bc -l << EOF
scale=4
$RCRrt + $rtuse
EOF
)
RCRwt=$( bc -l << EOF
scale=4
$RCRwt + $wtuse
EOF
)
done
echo " RESOURCES CURRENTLY RUNNING:"
echo "------------------------------"
echo " Number of running jobs : "$numl" ($tnodes nodes)"
echo " Resources in play (used/request): $RCRrt / $RCRwt nodedays"
echo " "
#step two queued resources
RCQwt="0"
numl=`qstat -n | grep " Q " | wc -l`
for (( i=1 ; i<= $numl ; i++ )); do
lin=`qstat -n | grep " Q " | head -$i | tail -1`
nodes=`echo $lin | awk '{print $6}'`
lin=`echo $lin | sed 's/:/\ /g'`
wth=`echo $lin | awk '{print $9}'`
wtm=`echo $lin | awk '{print $10}'`
wts=`echo $lin | awk '{print $11}'`
wtuse=$( bc -l << EOF
scale=4
$nodes * (($wth + ( $wtm + ( $wts / 60.0000 ) )/60.0000)/24.00)
EOF
)
RCQwt=$( bc -l << EOF
scale=4
$RCQwt + $wtuse
EOF
)
done
echo " RESOURCES CURRENTLY QUEUED:"
echo "------------------------------"
echo " Number of queued jobs : "$numl
echo " Resources requested : $RCQwt nodedays"
echo " "
#step three used resources in ended jobs
cnt="0"
RCFrt="0"
RCFwt="0"
numl=`qstat | grep " C " | wc -l`
#mam-statement does give the ID's but no further info can be extracted from qstat.
#numl=`mam-statement | grep $project | grep 'UsageRecord' | wc -l`
# list of jobIDs
LST=`qstat | grep " C " | sed 's/\./\ /g' | awk '{print $1}'`
#LST=`mam-statement | grep $project | grep 'UsageRecord' | awk '{print $3}'`
for jobid in `echo $LST`; do
#check if the job actually ran
tst=`qstat -f $jobid | grep 'walltime' | wc -l`
if [ "$tst" == "2" ]; then
nodes=`qstat -f $jobid | grep nodect | awk '{print $3}'`
lin=`qstat -f $jobid | grep 'resources_used.walltime' | awk '{print $3}' | sed 's/:/\ /g'`
rth=`echo $lin | awk '{print $1}'`
rtm=`echo $lin | awk '{print $2}'`
rts=`echo $lin | awk '{print $3}'`
lin=`qstat -f $jobid | grep 'Resource_List.walltime' | awk '{print $3}' | sed 's/:/\ /g'`
wth=`echo $lin | awk '{print $1}'`
wtm=`echo $lin | awk '{print $2}'`
wts=`echo $lin | awk '{print $3}'`
rtuse=$( bc -l << EOF
scale=4
$nodes * (($rth + ( $rtm + ( $rts / 60.0000 ) )/60.0000)/24.00)
EOF
)
wtuse=$( bc -l << EOF
scale=4
$nodes * (($wth + ( $wtm + ( $wts / 60.0000 ) )/60.0000)/24.00)
EOF
)
RCFrt=$( bc -l << EOF
scale=4
$RCFrt + $rtuse
EOF
)
RCFwt=$( bc -l << EOF
scale=4
$RCFwt + $wtuse
EOF
)
else
# if the job didn't run
cnt=$(bc -l << EOF
scale=0
$cnt + 1
EOF
)
fi
done
pct=$( bc -l << EOF
scale=3
( $RCFrt / $RCFwt ) * 100.00
EOF
)
#remove the number of not-run jobs
numl=$(bc -l << EOF
scale=0
$numl - $cnt
EOF
)
echo " RESOURCES RECENTLY COMPLETED JOBS:"
echo "-------------------------------------"
echo " Number of jobs not run : "$cnt
echo " Number of compled jobs : "$numl
echo " Resources used : $RCFrt / $RCFwt nodedays"
echo " This is "$pct" % of the requested resources."
echo " "
# finish with the Balance
mam-balance
[/codesyntax]
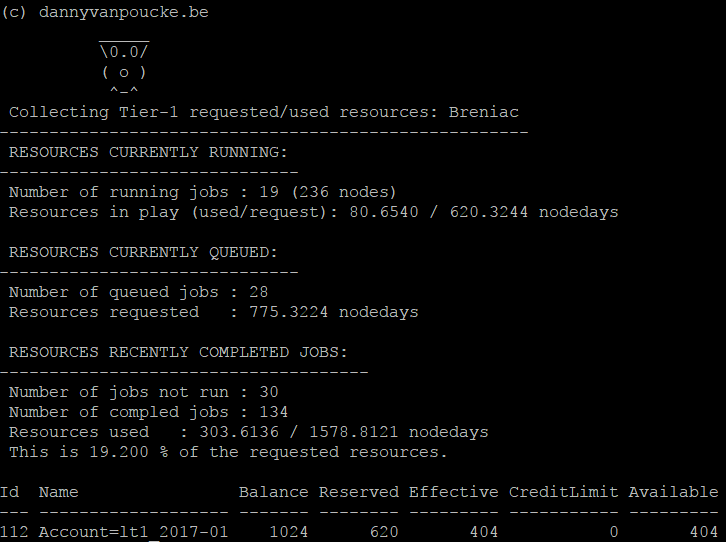
Output of the Bash Script.
Currently, the last part, on the completed jobs, only provides data based on the most recent jobs. Apparently the full qstat information of older jobs is erased. However, it still provides an educated guess of what you will be using for the still queued jobs.
Permanent link to this article: https://dannyvanpoucke.be/pbsbalancecollector-en/