As a computational materials scientist with a main research interest in the ab initio simulation of materials, computational resources are the life-blood of my research. Over the last decade, I have seen my resource usage grow from less than 100.000 CPU hours per year to several million CPU-hours per year. To satisfy this need for computational resources I have to make use of HPC facilities, like the TIER-2 machines available at the Flemish universities and the Flemish TIER-1 supercomputer, currently hosted at KU Leuven. At the international level, computational scientists have access to so called TIER-0 machines, something I no doubt will make use of in the future. Before I continue, let me first explain a little what this TIER-X business actually means.
The TIER-X notation is used to give an indication of the size of the computer/supercomputer indicated. There are 4 sizes:
- TIER-3: This is your personal computer(laptop/desktop) or even a small local cluster of a research group. It can contain from one (desktop) up to a few hundred CPU’s (local cluster). Within materials research, this is sufficient for quite a few tasks: post-processing of data, simple force-field based calculations, or even small quantum chemical or solid state calculations. A large fraction of the work during my first Ph.D. was performed on the local cluster of the CMS.
- TIER-2: This is a supercomputer hosted by an institute or university. It generally contains over 1000 CPUs and has a peak performance of >10 TFLOPS (1012 Floating Point Operations Per Second, compare this to 1-50×109 FLOPS or 1-25 GFLOPS of an average personal computer). The TIER-2 facilities of the VUB and UAntwerp both have a peak performance of about 75TFLOPS , while the machines at Ghent University and the KU Leuven/Uhasselt facilities both have a peak performance of about 230 TFLOPS. Using these machines I was able to perform the calculations necessary for my study of dopant elements in cerates (and obtain my second Ph.D.).
- TIER-1: Moving up one more step, there are the national/regional supercomputers. These generally contain over 10.000 CPUs and have a peak performance of over 100 TFLOPS. In Flanders the Flemish Supercomputer Center (VSC) manages the TIER-1 machine (which is being funded by the 5 Flemish universities). The first TIER-1 machine was hosted at Ghent University, while the second and current one is hosted at KU Leuven, an has a peak performance of 623 TFLOPS (more than all TIER-2 machines combined), and cost about 5.5 Million € (one of the reasons it is a regional machine). Over the last 5 years, I was granted over 10 Million hours of CPU time, sufficient for my study of Metal-Organic Frameworks and defects in diamond.
- TIER-0: This are international level supercomputers. These machines contain over 100.000 CPUs, and have a peak performance in excess of 1 PFLOP (1 PetaFLOP = 1000 TFLOPS). In Europe the TIER-0 facilities are available to researchers via the PRACE network (access to 7 TIER-0 machines, accumulated 43.49 PFLOPS).
This is roughly the status of what is available today for Flemish scientists at various levels. With the constantly growing demand for more processing power, the European union, in name of EuroHPC, has decided in march of this year, that Europe will host two Exa-scale computers. These machines will have a peak performance of at least 1 EFLOPS, or 1000 PFLOPS. These machines are expected to be build by 2024-2025. In June, Belgium signed up to EuroHPC as the eighth country participating, in addition, to the initial 7 countries (Germany, France, Spain, Portugal, Italy, Luxemburg and The Netherlands).
This is very good news for all involved in computational research in Flanders. There is the plan to build these machines, there is a deadline, …there just isn’t an idea of what these machines should look like (except: they will be big, massively power consuming and have a target peak performance). To get an idea what users expect of such a machine, Tier-1 and HPC users have been asked to put forward requests/suggestions of what they want.
From my user personal experience, and extrapolating from my own usage I see myself easily using 20 million hours of CPU time each year by the time these Exa-scale machines are build. Leading a computational group would multiply this value. And then we are talking about simple production purpose calculations for “standard” problems.
The claim that an Exa-scale scale machine runs 1000x faster than a peta-scale machine, is not entirely justified, at least not for the software I am generally encountering. As software seldom scales linearly, the speed-gain from Exa-scale machinery mainly comes from the ability to perform many more calculations in parallel. (There are some exceptions which will gain within the single job area, but this type of jobs is limited.) Within my own field, quantum mechanical calculation of the electronic structure of periodic atomic systems, the all required resources tend to grow with growth of the problem size. As such, a larger system (=more atoms) requires more CPU-time, but also more memory. This means that compute nodes with many cores are welcome and desired, but these cores need the associated memory. Doubling the cores would require the memory on a node to be doubled as well. Communication between the nodes should be fast as well, as this will be the main limiting factor on the scaling performance. If all this is implemented well, then the time to solution of a project (not a single calculation) will improve significantly with access to Exa-scale resources. The factor will not be 100x from a Pflops system, but could be much better than 10x. This factor 10 also takes into account that projects will have access to much more demanding calculations as a default (Hybrid functional structure optimization instead of simple density functional theory structure optimization, which is ~1000x cheaper for plane wave methods but is less accurate).
At this scale, parallelism is very important, and implementing this into a program is far from a trivial task. As most physicists/mathematicians/chemists/engineers may have the skills for writing scientifically sound software, we are not computer-scientists and our available time and skills are limited in this regard. For this reason, it will become more important for the HPC-facility to provide parallelization of software as a service. I.e. have a group of highly skilled computer scientists available to assist or even perform this task.
Next to having the best implementation of software available, it should also be possible to get access to these machines. This should not be limited to a happy few through a peer review process which just wastes human research potential. Instead access to these should be a mix of guaranteed access and peer review.
- Guaranteed access: For standard production projects (5-25 million CPU hours/year) university researchers should have a guaranteed access model. This would allow them to perform state of the are research without too much overhead. To prevent access to people without the proven necessary need/skills it could be implemented that a user-database is created and appended upon each application. Upon first application, a local HPC-team (country/region/university Tier-1 infrastructure) would have to provide a recommendation with regard to the user, including a statement of the applicant’s resource usage at that facility. Getting resources in a guaranteed access project would also require a limited project proposal (max 2 pages, including user credentials, requested resources, and small description of the project)
- Peer review access: This would be for special projects, in which the researcher requires a huge chunk of resources to perform highly specialized calculations or large High-throughput exercises (order of 250-1000 million CPU hours, e.g. Nature Communications 8, 15959 (2017)). In this case a full project with serious peer review (including rebuttal stage, or the possibility to resubmit after considering the indicated problems). The goal of this peer review system should not be to limit the number of accepted projects, but to make sure the accepted projects run successfully.
- Pay per use: This should be the option for industrial/commercial users.
What could an HPC user as myself do to contribute to the success of EuroHPC? This is rather simple, run the machine as a pilot user (I have experience on most of the TIER-2 clusters of Ghent University and both Flemish Tier-1 machines. I successfully crashed the programs I am using by pushing them beyond their limits during pilot testing, and ran into rather unfortunate issues. 🙂 That is the job of a pilot user, use the machine/software in unexpected ways, such that this can be resolved/fixed by the time the bulk of the users get access.) and perform peer review of the lager specialized projects.
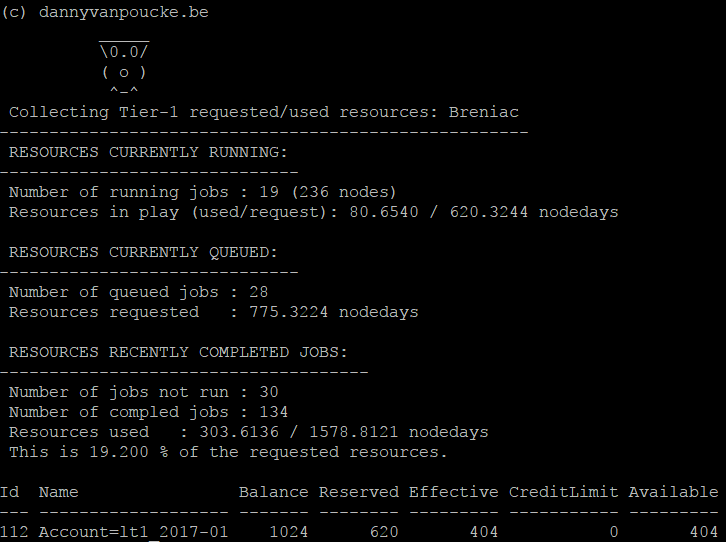
Now the only thing left to do is wait. Wait for the Exa-scale supercomputers to be build…7 years to go…about 92 node-days on Breniac…a starting grant…one long weekend of calculations.
Appendix
For simplicity I use the term CPU to indicate a single compute core, even though technically, nowadays a single CPU will contain multiple cores (desktop/laptop: 2-8 cores, HPC-compute node: 2-20 cores / CPU (or more) ). This to make comparison a bit more easy.
Furthermore, modern computer systems start more and more to rely on GPU performance as well, which is also a possible road toward Exa-scale computing.
Orders of magnitude:
- G = Giga = 109
- T = Tera = 1012
- P = Peta = 1015
- E = Exa = 1018



 Today is a joyful day, as I am getting married to my
Today is a joyful day, as I am getting married to my